Marta Kempf  ,
Monika Konnert
,
Monika Konnert
Distribution of genetic diversity in Fagus sylvatica at the north-eastern edge of the natural range
Kempf M., Konnert M. (2016). Distribution of genetic diversity in Fagus sylvatica at the north-eastern edge of the natural range. Silva Fennica vol. 50 no. 4 article id 1663. https://doi.org/10.14214/sf.1663
Highlights
- European beech at the north-eastern edge of its natural range in Poland have a high level of genetic variation, similar to the populations from Central Europe
- The differences between the beech provenances from the two centres in Poland, which were previously identified on the basis of pollen analyses and phenotypic traits, have now been genetically confirmed.
Abstract
An understanding of the genetic variation of the beech, especially at the edge of its natural distribution, is essential because of the change in natural distribution of the species resulting from changing climatic conditions. The main aim of the study was to determine the level of genetic diversity of European beech at the north-eastern edge of its natural range. The other aim was to check the genetic variation of beech from the two centres, the north and the south of Poland, which were identified in previous findings based on pollen analyses and phenotypic traits. The research material was the progeny of twelve beech provenances. The genetic structure of the populations was determined by ten highly variable microsatellite DNA loci. The results confirmed the high genetic diversity of beech at the north-eastern edge of its natural distribution, which infers the probability of their good adaptation to the changing climate and an extension of the range. Genetic analyses confirmed the existence of two genetic centres for beech in Poland. The populations from south-eastern Poland had a slightly higher diversity than the populations from the north-western area, which may indicate that the colonisation of Poland occurred by two routes. The results are important for creating the borders of the provenance regions and for limiting the transfer of seeds and seedlings. The choice of forest reproductive material, based on the knowledge of genetic diversity, is very important for the stability of future forests.
Keywords
Poland;
nSSR markers;
European beech;
limit of natural distribution
-
Kempf,
Department of Genetics and Forest Tree Breeding, Faculty of Forestry, University of Agriculture in Krakow, Al. 29-listopada 46, 31–425 Krakow
E-mail
m.kempf@ur.krakow.pl
- Konnert, Bavarian Office for Forest Seeding and Planting, Forstamtsplatz 1, 83317 Teisendorf, Germany E-mail monika.konnert@asp.bayern.de
Received 10 June 2016 Accepted 18 August 2016 Published 26 August 2016
Views 209313
Available at https://doi.org/10.14214/sf.1663 | Download PDF

1 Introduction
The European beech (Fagus sylvatica L.) is one of the most widely found broadleaved tree species in southern, central and western Europe. Its range is from Sicily in the south to southern Scandinavia in the north and from northeastern Spain in the west to the Carpathian Mountains in the east (Hulten and Fries 1986). The north eastern edge of the natural distribution of beech is in Poland (Latałowa et al. 2004). The great adaptive potential of the European beech enables it to flourish in a wide range of natural conditions and so it plays an important economic and ecological role. In view of climate change, the study of genetic variation and differentiation of beech populations is crucial for determining the adaptive potential of the species. This is especially important for the populations situated at the edges of the natural distribution, which in light of the predicted climate change, will play a significant role in the dispersal of the species in the north-easterly direction (Matuszkiewicz 2001; Latałowa 2004). Identification of the genetic and demographic processes is essential for determining priorities in the management of genetic resources (Hampe and Petit 2005), including the legal regulations associated with the limits of transfer of forest reproductive material (FRM). The application of the knowledge gained will be of essential importance for the health and future of forests.
The European distribution model of the genetic diversity of trees illustrates the populations’ evolutionary history (Leonardi and Menozzi 1995). The observed variation reflects the migration processes, including the inter-mixing of populations with a different evolutionary history. To correctly interpret the genetic diversity of the population of forest trees, inter alia, the number of refugial areas of the species and the routes of its post-glacial migration (Widmer and Lexer 2001) should be taken into account. The original model of the distribution of genetic diversity assumed that it was a result of the long-term isolation of populations during the last glacial period. The refugial areas, located in the south of Europe, were perceived as the places storing genetic richness, which was decreasing with the increasing distance from the distribution centres of a particular species, so called “southern richness and northern purity” (Hewitt 1999). Currently the refugial areas located in the south of Europe are considered as the sources of allele richness, but not as sources of genetic diversity (Widmer and Lexer 2001; Petit et al. 2003). According to new opinions in Central and Western Europe there could be an unrecognised local microrefugia, constituting, to date, an underestimated source of variation (Willis 2000; Mitka et al. 2014). The recent research shows that the history of post-glacial migration of many forest trees is much more complex than the explanation provided by previous hypotheses (Stewart and Lister 2001; Keir et al. 2011; De Lafontaine et al. 2013). Frequently, the process of species migration was controlled by the local conditions and therefore its dynamics in a region varied (Ralska-Jasiewiczowa et al. 2003; Magri et al. 2006).
For beech the most important period in its recolonisation history was the last interglacial era (Magri et al. 2006). Beech entered Poland in the subboreal period, first from the south and south-west into the Carpathian region – probably via the Beskid Niski Mountains and the Moravian Gate (Szafer 1932; Latałowa 1992). The second expansion route was from the northwest through the lakes of Western Pomerania, from where beech spread further to the east and the south (Ralska-Jasiewiczowa 1983; Giesecke et al. 2007; Magri et al. 2008).
As pollen data show there are clearly two centres of this species in Poland, one in the north-west and the second in the south-east (Latałowa et al. 2004). The differences between the capacity of beech from the northwest and southeast of Poland to adapt and grow have been confirmed by many years of research on interspecies variability in provenance experiments (Barzdajan and Rzeźnik 2002).
This paper presents the results of nuclear-microsatellite analyses of beech populations from the two Polish regions with a focus on the following questions:
Is Fagus sylvatica in Poland structured into a northern and a southern genetic group? Is the genetic diversity of beech in the two regions significantly different and is this difference genetically justified?
Does Fagus sylvatica at the north-eastern edge of its natural range exhibit high or low genetic diversity?
2 Material and methods
We investigated the progenies of twelve beech provenances from northern and southern Poland (Table 1 and Fig. 1) in an experimental plot in Krynica (49°24´N, 20°55´E). The experimental plot was established in 1995 with provenances of beech from the limit of its natural range in Poland. The collection of seeds to establish the project was made to achieve a minimum of 50 trees per stand. The detailed characteristics of the experimental plot and the provenances were presented in an earlier paper (Sabor and Żuchowska 2001). Leaves for DNA analysis were collected from approximately 46 trees per provenances. The trees were 22 years old.
| Table 1. Description of investigated provenances. | ||||||
| Ecotypes | No. of prov. | Provenance name | Altitude | Geographic coordinates | Age of adult stand | |
| latitude | longitude | |||||
| northern | 3 | Radachowo | 193 | 53°05´N | 15°54´E | 107 |
| 7 | Gniewino | 115 | 54°37´N | 18°18´E | 110 | |
| 8 | Jelenino | 130 | 53°40´N | 16°40´E | 96 | |
| 9 | Dalęcino | 130 | 53°45´N | 16°30´E | 111 | |
| 11 | Marianowo | 70 | 54°23´N | 18°31´E | 105 | |
| 17 | Mikołajki | 40 | 53°51´N | 19°10´E | 159 | |
| 19 | Tumiany | 175 | 53°58´N | 20°57´E | 130 | |
| southern | 31 | Pokrzywna | 425 | 50°20´N | 17°20´E | 130 |
| 34 | Jeleniów | 350 | 50°50´N | 20°59´E | 101 | |
| 37 | Marynin | 170 | 50°23´N | 22°17´E | 96 | |
| 39 | Bukowa | 550 | 49°39´N | 18°54´E | 122 | |
| 42 | Moczarna | 700–920 | 49°07´N | 22°29´E | 135 | |
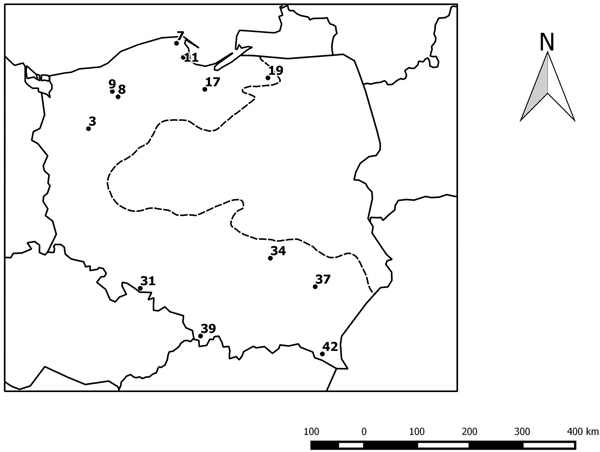
Fig. 1. Geographical distribution of the studied provenances. The black circles indicate the location of the beech provenances, see Table 1. The dotted line indicates the limit of the beech range in Poland.
2.1 DNA analysis
The extraction of genomic DNA from leaves was undertaken by a modified ATMAP method (Dumolin et al. 1995). The quantity and quality of the isolated DNA was assessed using a spectrophotometer (GeneQuant pro, Amersham Biosciences). All individuals were genotyped at ten highly variable primer pairs of polymorphic nuclear microsatellite loci: FS3-04, FS1-15 (Pastorelli et al. 2003), DE576, csolfagus19, csolfagus31 (Lefèvre et al. 2012), mfc5, mfc7, mfc11 (Tanaka et al. 1999), sfc0036 (Asuka et al. 2004) and mfs11 (Vornam et al. 2004). A PCR (Polymerase Chain Reaction) reaction was undertaken using primers labelled with different fluorescent dyes: Cy5 for loci DE576A0, FS1-15, mfc11, mfs11; Dy751 for csolf19, mfc7 and IRD700 for csolf31, FS3-04, mfc5 and sfc0036.
PCR amplifications were conducted in a solution of total volume 15 µl containing 7.5 µl Qiagen Multiplex PCR Master Mix 2x, 5 µl H2O RNnase-free, 1.5 µl Primer Mix and 1 µl template DNA. The PCR program, the same for all loci, was started employing an initial denaturation at 95 °C for 15 min, followed by 24 cycles of 94 °C for 30 sec, 57 °C for 90 sec, 72 °C for 30 sec and a final elongated step of 15 minutes at 60 °C. The determination of fragment length and allele assignment were undertaken using the fragment analysis tool of an automated sequencer CEQ8000 (Beckman-Coulter; GeneMapper® program Version 4.0). An internal size standard (LIZ500) was used for sizing the PCR fragments.
2.2 Data analysis
The frequencies of null alleles (non-amplifiable fragments) per locus and per population were estimated using the Microchecker software (Van Oosterhout et al. 2004). The adjusted frequencies of null alleles according to Van Oosterhout et al. (2006) were used for further estimating the genetic variation. Genetic diversity within populations was estimated on the basis of the mean number of alleles per locus (NA), the effective number of alleles (NE), the observed and the expected heterozygosity (HO and HE). To calculate these parameters the software GenAlEx 6.41 (Peakall and Smouse 2006) was used. FSTAT 2.9.3.2 software (Goudet 1995) was used to calculate the inbreeding coefficients (FIS) for each locus. The significance was tested by performing 10 000 randomisations of alleles among individuals within the samples. The genetic differentiation patterns among populations were detected by calculating the populations pairwise FST (Weir and Cockerham 1984). The statistical significance was tested by using 10 000 permutations of individuals between the compared populations. To quantify the distribution of total variance at different hierarchical levels, an analysis of the molecular variance (AMOVA) was carried out using GenAlEx 6.41 (Peakall and Smouse 2006). The same software was used for a Mantel test, to investigate the relationship between the pairwise population FST values and geographic distance. The correlation between the geographic coordinates (longitude, latitude) and the genetic diversity parameters were studied. A principal coordinate analysis (PCoA) was used to group populations according to their geographical origin based on pairwise FST values (Hartl and Clark 1997). The Bayesian clustering method implemented in the software STRUCTURE 2.3.4 (Pritchard et al. 2000; Falush et al. 2003; Falush et al. 2007; Hubisz et al. 2009) was used to detect the genetic differentiation of populations. The multilocus SSR data of all individuals were used to calculate posteriori probabilities of membership and to define the K subpopulations based on their genetic similarity. The admixture model and correlated allele frequencies were used with no prior information. Five independent runs, each with 50 000 MCMC iterations after 10 000 burn-in periods, were carried out for the K set between 1 and 15. The ∆K statistics (Evanno et al. 2005) were used to detect the uppermost hierarchical level of the population structure, based on the rate of change between successive K values. The best estimate of K (∆K) was calculated using the web-based STRUCTURE HARVESTER program (Earl and von Holdt 2012). After identifying the optimal K by this method, samples were placed into the cluster for which they showed the highest percentage of contribution.
3 Results
All ten microsatellite loci used in this study were polymorphic and displayed a high number of alleles and a wide range of PCR products. A total of 132 alleles were detected.
The total number of alleles per locus ranged from 5 (FS3-04) to 25 (mfc5), with an average of 13.2 (Table 2). The highest number of alleles (mean value, NA) and the highest effective number of alleles (NE) were observed for the locus mfc5 with 16.1 and 8.09, respectively. The observed heterozygosity (HO) ranged from 0.347 (Fs3-04) to 0.837 (csolf31), with an average of 0.698. The mean expected heterozygosity (HE) was 0.701, and ranged from 0.346 (Fs3-04) to 0.866 (mfc5). In all analysed populations private alleles were found. The frequencies of these alleles were low, ranging between 0.01 and 0.13, with an average of 0.03. The values of the inbreeding coefficients (FIS) were near zero in most cases. This indicates that there was no excess of homo- and heterozygotes in comparison to the expected values. However, significant and high FIS values were observed for two loci, indicating a deficiency of heterozygotes (Fs3-04 and mfc5). The genetic differentiation (FST) between populations averaged 0.041. It varied from 0.030 to 0.087 at locus FS3-04, a very high value compared to the other nine loci.
| Table 2. Summary statistics for the 10 microsatellite loci. | |||||||||
| Locus | Size range (bp) | No. of alleles | NA | NE | HO | HE | FIS | FST | |
| csolf31 | 106–128 | 13 | 10.40 | 5.51 | 0.837 | 0.815 | –0.017 | 0.030 | |
| FS3-04 | 193–205 | 5 | 3.60 | 1.61 | 0.347 | 0.346 | 0.034 | 0.087 | |
| mfc11 | 321–355 | 12 | 7.70 | 4.03 | 0.738 | 0.739 | –0.010 | 0.040 | |
| FS1-15 | 92–146 | 23 | 9.80 | 5.52 | 0.762 | 0.813 | 0.055*** | 0.034 | |
| csolf19 | 151–181 | 12 | 9.70 | 4.87 | 0.795 | 0.789 | –0.004 | 0.032 | |
| mfs11 | 128–164 | 12 | 7.90 | 4.06 | 0.731 | 0.740 | 0.001 | 0.041 | |
| mfc7 | 111–173 | 15 | 5.90 | 2.13 | 0.526 | 0.501 | –0.045 | 0.040 | |
| mfc5 | 277–333 | 25 | 16.10 | 8.09 | 0.800 | 0.866 | 0.071*** | 0.037 | |
| sfc0036 | 97–111 | 8 | 7.10 | 4.13 | 0.758 | 0.747 | –0.013 | 0.030 | |
| DE576A0 | 219–240 | 7 | 5.30 | 2.92 | 0.683 | 0.651 | –0.040 | 0.037 | |
| Mean (overall) | - | 13.2 | 8.35 | 4.29 | 0.698 | 0.701 | 0.003 | 0.041 | |
| The size range: the minimum and maximum lengths of PCR product detected for a locus (in base pairs), NA – mean number of alleles, NE – effective number of alleles, HO – observed hetrozygosity, HE – expected hetrozygosity, FIS – inbreeding coefficient. Significance of FIS values: *** p<0.001 | |||||||||
For the 12 provenances the mean number of alleles (NA) varies from 7.7 in provenance 8 Jelenino to 9.2 in provenance 39 Bukowa with a mean of 8.3 (Table 3). The effective number of alleles (NE) ranges from 3.52 (37 Marynin) to 4.88 (31 Pokrzywna). Generally, slightly higher values of NA and NE were found in the southern provenances. The observed heterozygosity (HO) was the highest (0.750) in population 39 Bukowa and the lowest (0.673) in population 37 Marynin with the mean of 0.701. The expected heterozygosity (HE) ranged from 0.665 (37 Marynin) to 0.730 (39 Bukowa). The mean expected heterozygosity was 0.704. Most provenances showed a lack of significant deviations from the expected Hardy-Weinberg frequencies. The mean value of the fixation index was 0.004. In most populations there were more homozygous genotypes than expected (F positive). Provenances 34 Jeleniów, 37 Marynin and 39 Bukowa, all from the south, showed significantly higher heterozygosity than expected.
| Table 3. Diversity measures in Fagus sylvatica. | |||||||||
| Ecotypes | Provenance | NA | NE | HO | HE | F | No. Private Alleles | Freq | |
| northern | 3 | Radachowo | 7.90 | 4.11 | 0.677 | 0.694 | 0.023 | 1 | 0.02 |
| 7 | Gniewino | 8.00 | 3.81 | 0.707 | 0.707 | –0.003 | 3 | 0.06 | |
| 8 | Jelenino | 7.70 | 4.49 | 0.679 | 0.698 | 0.025* | 1 | 0.02 | |
| 9 | Dalęcino | 7.80 | 4.32 | 0.707 | 0.710 | 0.010 | 1 | 0.01 | |
| 11 | Marianowo | 8.30 | 4.09 | 0.681 | 0.696 | 0.011 | 1 | 0.01 | |
| 17 | Mikołajki | 8.10 | 4.25 | 0.715 | 0.723 | 0.014* | 2 | 0.02 | |
| 19 | Tumiany | 8.00 | 4.39 | 0.715 | 0.717 | 0.017 | 1 | 0.01 | |
| Mean | 7.97 | 4.21 | 0.697 | 0.706 | 0.014 | 0.02 | |||
| southern | 31 | Pokrzywna | 9.00 | 4.88 | 0.679 | 0.696 | 0.038 | 3 | 0.03 |
| 34 | Jeleniów | 8.30 | 4.56 | 0.735 | 0.710 | –0.041* | 2 | 0.02 | |
| 37 | Marynin | 8.20 | 3.52 | 0.673 | 0.665 | –0.015* | 1 | 0.02 | |
| 39 | Bukowa | 9.20 | 4.60 | 0.750 | 0.730 | –0.041* | 6 | 0.13 | |
| 42 | Moczarna | 9.10 | 4.48 | 0.688 | 0.701 | 0.014 | 4 | 0.04 | |
| Mean | 8.76 | 4.41 | 0.705 | 0.701 | –0.009 | 0.05 | |||
| Mean | 8.30 | 4.29 | 0.701 | 0.704 | 0.004 | 0.03 | |||
| NA – mean number of alleles, NE – effective number of alleles, HO – observed heterozygosity, HE – expected heterozygosity, F – fixation index. Significance of F values: * p<0.01, number of private alleles and their frequency (Freq). | |||||||||
The ‘Analysis of Molecular Variance’ (AMOVA), that allows the partitioning of variation among and within populations, indicated that variance was highest within populations (93%) (Table 4). A variation of about 5% of among populations occurred within regions, while only 2% variation was found between the regions.
| Table 4. Hierarchical analysis of molecular variance (AMOVA) based on allelic distance matrix. | ||||||
| Variance component | df | SS | MS | Variance | % Total | P |
| Among Regions | 1 | 70.486 | 70.486 | 0.162 | 2% | 0.010 |
| Among Pops | 10 | 250.160 | 25.016 | 0.368 | 5% | 0.010 |
| Within Pops | 564 | 4134.229 | 7.330 | 7.330 | 93% | 0.010 |
| Total | 575 | 4454.875 | ||||
| df – degree of freedom, SS – Sum of squares, MS – Mean squares, P – probability | ||||||
The Mantel test (Fig. 2) showed a positive and significant correlation coefficient between the geographic distance and pairwise FST values (R = 0.721, p = 0.01). The existence of a distinct geographical structure of the studied populations was indicated. Significant, but negative, correlation with latitude was found only for the mean number of alleles NA (R = –0.812, p < 0.01). A longitudinal trend of genetic variation was not detected.
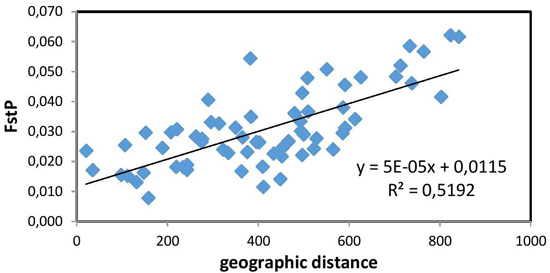
Fig. 2. Relationship between pairwise FST values and geographic distance (km) (R = 0.721, p = 0.01) between the studied 12 populations of European beech.
The results of the principal coordinate analysis (PCoA) based on pairwise FST are presented in Fig. 3. The first two coordinates explain 47% of the total variation. Coordinate 1 indicates the separation of the northern and southern beech populations. One population from the south (31 Pokrzywna) was included in the group of northern populations. The spatial differentiation into a northern and a southern group was confirmed by the STRUCTURE analysis.
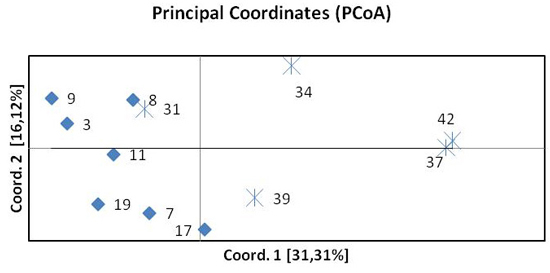
Fig. 3. Principal Coordinates Analysis (PCoA) of pairwise FST of twelve beech populations. Diamonds represent provenances from the north, crosses provenances from the south.
All loci were jointly used for calculating K subpopulations based on the membership proportions. The rate of change of data posterior probability (lnP(D)) between successive runs was the highest for K = 3 (Fig. 4). The clusters were not clearly distinguished. Admixture of clusters in the south and north provenances was high, but a trend of differentiation could be detected. The south populations are more represented by the green cluster and the north populations by the blue cluster. The average share of the green cluster in the southern provenances was 55%, whereas in the north it was only 22%. For the northern populations the blue cluster averaged 43%, whereas in the south only 19%. The red cluster was characteristic for all provenances, averaging 34%. Two subpopulations were indicated by the STRUCTURE analysis, corresponding to the two groups of populations given by the PCoA analysis. The populations from the south form a clearly differentiated group. Only the 31 Pokrzywna is similar to the populations in the north.
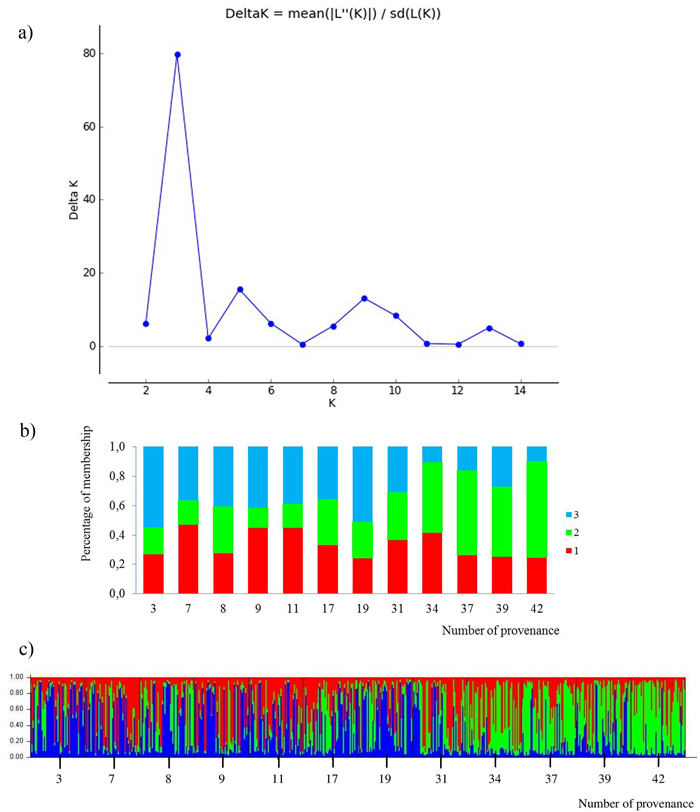
Fig. 4. Results for STRUCTURE analysis:
a) values of the statistic “Delta K” for all calculated K-values from 1–15 based on the combined dataset of nuclear microsatellites. The highest “Delta K” corresponds to the uppermost level of hierarchy among runs (Evanno et al. 2005).
b) proportion of membership of each individual to three assumed subpopulations (K = 3).
c) proportion of membership of each individual to three assumed subpopulations (K = 3). (Northern provenances: 3 to 19; Southern provenances 31 to 42. For location names see Table 3). View larger in new window/tab.
4 Discussion
In this research the genetic diversity of twelve provenances of Fagus sylvatica from north-eastern part of its natural distribution range in Poland was studied. The analysis showed a clear genetic differentiation between provenances. The populations in the south-east had slightly higher variability (diversity, heterozygosity) than the populations in the north-west. In general the genetic diversity of the investigated populations was similar to populations of Fagus sylvatica from Central Europe, determined using the same marker type (nuclear microsatellites) Vornam et al. (2004): He = 0.765, Bilela et al. (2012): He = 0.740–0.792, Piotti et al. (2012): He = 0.728–0.810, and Dounavi et al. (2016) for EST microsatellites (He = 0.564–0.701) for German beech provenances. Lower values of genetic diversity (He = 0.617 resp. He = 0.614–0.632) for some German beech provenances have been reported Seifert (2011) and Rajendra et al. (2014). Differences between the observed and expected values of heterozygosity in the studied populations were small. According to Hartl and Clark (1997) this is a typical finding for a genetically stable population.
An equally low value of the fixation index was reported for beech by Lander et al. (2011) (FIS = 0.01–0.064) and Rajendra et al. (2014) (FIS = –0.003). However, generally higher FIS values have been reported in numerous isozyme studies for beech Comps et al. (1990): FIS = 0.065, Comps et al. (1991): FIS = 0.115, Leonardi and Menozzi (1995): FIS = 0.117, Harter et al. (2015): FIS = 0.02. The same applies to some nuclear microsatellite markers (Buiteveld et al. 2007: FIS = 0.224, Piotti et al. 2012: FIS = 0.139–0.233, Paffetti et al. 2012: FIS = 0.239). The positive value of fixation indices has been explained by a non-random micro-spatial structure due to mating preferences between neighbouring individuals (Comps et al., 1990; Merzeau et al. 1994; Shanjani et al. 2011). It is obvious that the values of the inbreeding coefficient depend, to a large extent, also on the selection of analysed loci. Thus, for example, microsatellite markers with a high frequency of zero alleles increase the FIS values (Buiteveld et al. 2007; Rajendra et al. 2014). In the reported study the mean FIS was close to zero, negative for the southern populations and slightly higher and positive for the northern populations. An increase of the inbreeding coefficient (FIS) in the northern direction has also been reported by Comps et al. (2001) and Rajendra et al. (2014).
In the study most of the genetic diversity was attributable to the stands themselves. There was a small, but significant differentiation of 5% between stands. This low differentiation between stands is in accordance with other studies on Fagus sylvatica using both isoenzymes and DNA markers (Leonardi and Menozzi 1995; Larsen 1996; Konnert and Henkel 1997; Comps et al. 2001; Gömöry et al. 2003; Buiteveld et al. 2007; Kraj and Sztorc 2009). A low, but statistically important (p = 0.01), 2% genetic differentiation between regions was found, which reflects the geographical patterns of genetic variation. Genetic analyses confirmed the existence of two genetic centres for beech in Poland, one in the northwest and another in the southeast. Thus the reported analysis confirms previous findings based on pollen analyses and phenotypic traits (adaptation features) (Barzdajan and Rzeźnik 2002; Latałowa et al. 2004). A similar, significant geographic division of populations, at similarly low FST values, was discovered earlier for beech in Italy (Leonardi and Menozzi 1995) and for beech from Austria, France, Germany, Italy and the Netherlands (Buiteveld et al. 2007).
Additionally, a continuous (clinal) increase in genetic diversity with increasing geographical separation was found, which confirms the work of Hampe and Petit (2005). The existence of a correlation between the genetic distance and geographical distance may provide evidence for a relationship between the genetic diversity and selective environmental gradients. This was the case in other studies using microsatellites with adaptive relevance (EST microsatellites) (Dounavi et al. 2016). However, it could also be a reflection of the hypothesis of genetic diversity based on different colonisation routes (Eckert et al. 2008). According to isoenzymes and DNA studies, a distinct variation in beech is found in the populations from various refugia, or from the various recolonisation routes after the ice age (Demesure et al. 1996; Gömöry et al. 1999; Comps et al. 2001; Magri et al. 2006). Originally it was assumed that beech in Central and Northern Europe arrived from the Balkan refugia (Huntley and Birks 1983; Taberlet et al. 1998). Currently the dominant opinion is that beech came to Central and Northern Europe from the refugia in southern France, the eastern Alps – Slovenia and Istria and probably from southern Moravia and southern Bohemia (Magri et al. 2006). The populations which survived the last glaciations in the Balkans and Italy were not the sources of today’s Central European beech (Magri et al. 2008). A significant role in the development of the diversity of European beech was played by the areas where the migration routes from different refugia interacted during the postglacial recolonisation (Comps et al. 2001; Widmer and Lexer 2001; Petit et al. 2003). Moreover, the latest paleoecological and phylogeographical research indicates the former existence of potential northern refugial areas, which survived the period of glaciations due to favourable climatic-habitat conditions (Willis and van Andel 2004; Tzedakis et al. 2013). So far, the role of such refugia in the analysis of the genetic variation within Europe has been underestimated (Willis and van Andel 2004; Svenning and Skov 2007). Western Slovakia and Northeastern Hungary are regarded as potential cryptorefugia for beech (Stewart and Lister 2001). Genetic differences between the northern and southern beech provenances found in this study may confirm that colonisation of Poland occurred by two routes. The first led from the Czech Republic, Moravia and the plains of Hungary around 3000 years BC, entering Poland from the south, through the Sudetes and Beskids Mountains (Ralska-Jasiewiczowa 1983; Ralska-Jasiewiczowa et al. 2003; Latałowa et al. 2004). The second led from Germany and colonised the north-western part of Poland (Ralska-Jasiewiczowa et al. 2003; Giesecke et al. 2007), from where it continued to the east and the south (Ralska-Jasiewiczowa 1983; Latałowa et al. 2004; Magri et al. 2006). A higher level of beech genetic diversity in south-eastern Poland may be evidence for the colonisation of this part of the country from the area of the Czech–Moravian lowlands and Hungary. The longer migration route of the northern beech populations led to their genetic depletion (Demesure et al. 1996; Comps et al. 2001). The evidence of this is the observed tendency of the decreasing genetic multiplicity, given as mean number of alleles (NA), from south to north, which closely corresponds to the reports of Comps et al. (2001) and Sułkowska (2010).
Climate change with increasing temperatures and dry periods will have an increasing impact on future beech stands (Fang and Lechowicz 2006). Results from modelling under different climate change scenarios predict a northward drift of the species range (Thuiller et al. 2006; Bellard et al. 2012; Falk and Hempelmann 2013) and the extinction of populations and a reduction of the range at the southern edge (Geßler et al. 2006; Dounavi et al. 2016). The climatic conditions in the north-eastern part of the present distribution range will become more favourable for beech. Actually beech is still in a phase of expansion as its range is showing further enlargement (Huntley and Birks 1983; Latałowa et al. 2004). Almost all prognoses state that the natural distribution range of the species will extend to southern and central Scandinavia, the Baltic countries and the larger part of Belarus (Thuiller et al. 2006; Bolte et al. 2010; Kramer et al. 2010). Analyses of climatic data suggest that the present conditions in north-eastern Poland should not be a limiting factor for the natural spread of beech into these areas (Matuszkiewicz 2001; Latałowa et al. 2004; Giesecke et al. 2007; Augustaitis et al. 2015). Due to the influence of climate change, the ecological, social and economic functions served by forests will depend on maintaining healthy, productive forest ecosystems, adjusted for the habitat (Chmura et al. 2010). In the near future the pool of species will probably not change drastically, but the frequency of their appearance, their growing performance, their seed production and resistance to external factors will change (Williams et al. 2012). Thus, the key issue is the implementation of management systems to fully maintain the diversity, while simultaneously maintaining the future stability of tree stands (Bolte et al. 2007). Therefore, forest management faces new challenges. The search for beech ecotypes having high adaptive potential in extreme growing conditions is essential to combat the predicted climate change, particularly the expected decrease of precipitation (Dounavi et al. 2016). An understanding of the genetic mechanisms which govern the physiological response of trees is very important. Research combining ecological and physiological approaches with assessments of tree growth in natural conditions is recommended (Geßler et al. 2006). The stands at the southern edge of the natural distribution range of the species will probably suffer drought stress (Netzer et al. 2016). Consequently, it is important for the genetic richness of these populations to be known, as it is assumed that greater genetic diversity provides greater possibilities for adaptation. A similar relationship exists at the northern edges of the natural distribution ranges. The populations at the northern leading edges, which in favourable conditions will be able to colonise new areas, should have a rich gene pool. All studied provenances showed a high genetic diversity, which implies the probability of good adaptation to the changing climate and the range being extended. The high adaptive capability of the Polish populations to stressful conditions compared to that of the German populations has already been confirmed by research (Rose et al. 2009).
An understanding of the genetic diversity of beech stands is also essential for activities associated with regeneration. Both natural and artificial regeneration are related to good seed years and good seed quality, but also with the management regime. Natural regeneration seems to permit an unrestricted flow of genetic information and thus the dynamic conservation of genetic multiplicity (Behm and Konnert 1990). For artificial regeneration, the choice of the appropriate FRM is of major importance. It is important to select provenances, not only on the basis of their adaptation value, but also on their genetic variability to provide the stability, needed for the generation of future forests. The present results complement the knowledge gained by multi-year observation of the variability of adaptation and growth features. They provide an insight into the genetic diversity and differentiation and demonstrate the high genetic variation of beech at the northern edge of its natural distribution.
The genetic differences between beeches from the two centres in Poland could result from their adaptation to local site conditions. The neutral molecular data could not be used as a surrogate for adaptive genetic information, but they provide partial insight into local adaptation or evolutionary potential (Kirk and Freeland 2011). The adaptation may lead to the genetic and phenotypic structuring of the population within the species, depending on the method of management over a few generations (Kramer et al. 2010). The local diversity of the beech population is frequently undervalued (Bolte et al. 2007) and without any doubt the maintenance of local diversity is important. However, to combat climate change a modest enrichment of populations from colder regions with provenances from more southern areas (Bilela et al. 2012) can be used to increase diversity and buffer against future climate uncertainty (Aitken and Bemmel 2015). To achieve this in a controlled way, the differences between the southern and northern provenances, indicated in the paper, should be maintained by the implementation of appropriate legal regulations.
Acknowledgements
Financial support by the Transnational Access to Research Infrastructures activity in the 7th Framework Programme of the EC under the Trees4Future project (no. 284181) for conducting the research is gratefully acknowledged.
References
Aitken S.N., Bemmels J.B. (2015). Time to get moving: assisted gene flow of forest trees. Evolutionary Applications 9(1): 271–90. http://dx.doi.org/10.1111/eva.12293.
Asuka Y., Tani N., Tsumura Y., Tomaru N. (2004). Development and characterization of microsatellite markers for Fagus crenata Blume. Molecular Ecology Notes 4(1): 101–103. http://dx.doi.org/10.1046/j.1471-8286.2003.00583.x.
Augustaitis A., Kliučius A., Marozas V., Pilkauskas M., Augustaitiene I., Staszewski T., Jansons A., Dreimanis A. (2015). Sensitivity of European beech trees to unfavorable environmental factors on the edge and outside of their distribution range in northeastern Europe. iForest – Biogeosciences and Forestry 9: 259–269. http://dx.doi.org/10.3832/ifor1398-008.
Barzdajan W., Rzeźnik Z. (2002). Wstępne wyniki międzynarodowego doświadczenia proweniencyjnego z bukiem (Fagus sylvatica L.) serii 1993/1995 w Leśnym Zakładzie Doświadczalnym Siemianice. [The preliminary results of the international provenance trial of common beech (Fagus sylvatica L.) of 1993/1995 series in Siemianice Experimental Forest District]. Sylwan 2(146): 149–164.
Behm A., Konnert M. (1999). Erhaltung forstlicher Genressourcen durch naturnahe Forstwirtschaft – eine reelle Chance? Mitteilungen Der Bundesforschungsanstalt Für Forst- Und Holzwirtschaft, Hamburg (194): 215–235.
Bellard C., Bertelsmeier C., Leadley P., Thuiller W., Courchamp F. (2012). Impacts of climate change on the future of biodiversity. Ecology Letters 15(4): 365–77. http://dx.doi.org/10.1111/j.1461-0248.2011.01736.x.
Bilela S., Dounavi A., Fussi B., Konnert M., Holst J., Mayer H., Rennenberg H., Simon J. (2012). Natural regeneration of Fagus sylvatica L. adapts with maturation to warmer and drier microclimatic conditions. Forest Ecology and Management 275: 60–67. http://dx.doi.org/10.1016/j.foreco.2012.03.009.
Bolte A., Czajkowski T., Kompa T., Birks H.J.B., Willis K.J. (2007). The north-eastern distribution range of European beech a review. Forestry 80(4): 413–429. http://dx.doi.org/10.1093/forestry/cpm028.
Bolte A., Hilbrig L., Grundmann B., Kampf F., Brunet J., Roloff A. (2010). Climate change impacts on stand structure and competitive interactions in a southern Swedish spruce–beech forest. European Journal of Forest Research 129(3): 261–276. http://dx.doi.org/10.1007/s10342-009-0323-1.
Buiteveld J., Vendramin G.G., Leonardi S., Kamer K., Geburek T. (2007). Genetic diversity and differentiation in European beech (Fagus sylvatica L.) stands varying in management history. Forest Ecology and Management 247(1–3): 98–106. http://dx.doi.org/10.1016/j.foreco.2007.04.018.
Chmura D.J., Howe G.T., Anderson P.D., Clair J.B.S. (2010). Przystosowanie drzew, lasów i leśnictwa do zmian klimatycznych. [Adaptation of trees, forests and forestry to climate change]. Sylwan 154(9): 587–602.
Comps B., Thiébaut B., Paule L., Merzeau D., Letouzey J. (1990). Allozymic variability in beechwoods (Fagus sylvatica L.) over central Europe: spatial differentiation among and within populations. Heredity 65(3): 407–417. http://dx.doi.org/10.1038/hdy.1990.111.
Comps B., Thiebaut B., Sugar I., Trinajstic I., Plazibat M. (1991). Genetic variation of the Croatian beech stands (Fagus sylvatica L.): spatial differentiation in connection with the environment. Annales Des Sciences Forestieres 48(1): 15–28. http://dx.doi.org/10.1051/forest:19910102.
Comps B., Gömöry D., Letouzey J., Thiébaut B., Petit R.J. (2001). Diverging trends between heterozygosity and allelic richness during postglacial colonization in the European beech. Genetics 157(1): 389–97.
De Lafontaine G., Ducousso A., Lefèvre S., Magnanou E., Petit R.J. (2013). Stronger spatial genetic structure in recolonized areas than in refugia in the European beech. Molecular Ecology 22(17): 4397–4412. http://dx.doi.org/10.1111/mec.12403.
Demesure B., Comps B., Petit R.J. (1996). Chloroplast DNA phylogeography of the common beech (Fagus sylvatica L.) in Europe. Evolution 50(6): 2515–2520. http://dx.doi.org/10.2307/2410719.
Dounavi A., Netzer F., Celepirovic N., Ivanković M., Burger J., Figueroa A.G., Schön S., Simon J., Cremer E., Fussi B., Konnert M., Rennenberg H. (2016). Genetic and physiological differences of European beech provenances (F. sylvatica L.) exposed to drought stress. Forest Ecology and Management 361: 226–236. http://dx.doi.org/10.1016/j.foreco.2015.11.014.
Dumolin S., Demesure B., Petit R.J. (1995). Inheritance of chloroplast and mitochondrial genomes in pedunculate oak investigated with an efficient PCR method. Theoretical and Applied Genetics 91(8): 1253–1256. http://dx.doi.org/10.1007/BF00220937.
Earl D.A., von Holdt B.M. (2012). STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conservation Genet Resour 4: 359–361. http://dx.doi.org/10.1007/s12686-011-9548-7.
Eckert C.G., Samis K.E., Lougheed S.C. (2008). Genetic variation across species’ geographical ranges: the central-marginal hypothesis and beyond. Molecular Ecology 17(5): 1170–1188. http://dx.doi.org/10.1111/j.1365-294X.2007.03659.x.
Evanno G., Regnaut S., Goudet J. (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Molecular Ecology 14(8): 2611–2620. http://dx.doi.org/10.1111/j.1365-294X.2005.02553.x.
Falk W., Hempelmann N. (2013). Species favourability shift in europe due to climate change: a case study for Fagus sylvatica L. and Picea abies (L.) Karst. based on an ensemble of climate models. Journal of Climatology vol. 2013, article 787250. 18 p. http://dx.doi.org/10.1155/2013/787250.
Falush D., Stephens M., Pritchard J.K. (2003). Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164(4): 1567–87.
Falush D., Stephens M., Pritchard J.K. (2007). Inference of population structure using multilocus genotype data: dominant markers and null alleles. Molecular Ecology Notes 7(4): 574–578. http://dx.doi.org/10.1111/j.1471-8286.2007.01758.x.
Fang J., Lechowicz M.J. (2006). Climatic limits for the present distribution of beech (Fagus L.) species in the world. Journal of Biogeography 33: 1804–1819. http://dx.doi.org/10.1111/j.1365-2699.2006.01533.x.
Geßler A., Keitel C., Kreuzwieser J., Matyssek R., Seiler W., Rennenberg H. (2006). Potential risks for European beech (Fagus sylvatica L.) in a changing climate. Trees 21(1): 1–11. http://dx.doi.org/10.1007/s00468-006-0107-x.
Giesecke T., Hickler T., Kunkel T., Sykes M.T., Bradshaw R.H.W. (2007). Towards an understanding of the Holocene distribution of Fagus sylvatica L. Journal of Biogeography 34(1): 118–131. http://dx.doi.org/10.1111/j.1365-2699.2006.01580.x.
Gömöry D., Paule L., Brus R., Zhelev P., Tomović Z., Gračan J. (1999). Genetic differentiation and phylogeny of beech on the Balkan peninsula. Journal of Evolutionary Biology 12(4): 746–754. http://dx.doi.org/10.1046/j.1420-9101.1999.00076.x.
Gömöry D., Paule L., Shvadchak I.M., Popescu F., Sułkowska M., Hynek V., Longauer R. (2003). Spatial patterns of the genetic differentiation in European beech (Fagus sylvatica L.) at allozyme loci in the Carpathians and the adjacent regions. Silvae Genetica 52(2): 78–83.
Goudet J. (1995). FSTAT (version 1.2): a computer program to calculate F-statistics. The Journal of Heredity 86(6): 485–486.
Hampe A., Petit R.J. (2005). Conserving biodiversity under climate change: the rear edge matters. Ecology Letters 8(5): 461–467. http://dx.doi.org/10.1111/j.1461-0248.2005.00739.x.
Harter D., Nagy L., Backhaus S., Beierkuhnlein C., Fussi B., Huber G., Jentsch A., Konnert M., Thiel D., Kreyling J. (2015). A comparison of genetic diversity and phenotypic plasticity among European beech (Fagus sylvatica L.) populations from Bulgaria and Germany under drought and temperature manipulation. International Journal of Plant Sciences 176(3): 232–244. http://dx.doi.org/10.1086/679349.
Hartl D.L., Clark A.G. (1997). Principles of Population Genetics. Sinauer Associates, Inc. Publishers Sunderland, Massachusetts. 652 p.
Hewitt G.M. (1999). Post-glacial re-colonization of European biota. Biological Journal of the Linnean Society 68(1–2): 87–112. http://dx.doi.org/10.1111/j.1095-8312.1999.tb01160.x.
Hubisz M.J., Falush D., Stephens M., Pritchard J.K. (2009). Inferring weak population structure with the assistance of sample group information. Molecular Ecology Resources 9(5): 1322–1332. http://dx.doi.org/10.1111/j.1755-0998.2009.02591.x.
Hultén E., Fries M. (1986). Atlas of North European vascular plants: north of the Tropic of Cancer. Koeltz Scientific Books, Königstein. Vols. I–III. 1172 p.
Huntley B., Birks H.J.B. (1983). An atlas of past and present pollen maps for Europe: 0–13 000 years ago. Cambridge University Press, Cambridge-London-New York-New Rochelle-Melbourne-Sydney. 667 p. http://dx.doi.org/10.1007/BF02902298.
Keir K.R., Bemmels J.B., Aitken S.N. (2011). Low genetic diversity, moderate local adaptation, and phylogeographic insights in Cornus nuttallii (Cornaceae). American Journal of Botany 98(8): 1327–1336. http://dx.doi.org/10.3732/ajb.1000466.
Kirk H., Freeland J.R. (2011). Applications and implications of neutral versus non-neutral markers in molecular ecology. International Journal of Molecular Sciences 12(6): 3966–3988. http://dx.doi.org/10.3390/ijms12063966.
Konnert M., Henkel W. (1997). Investigations on the genetic variation of beech (Fagus sylvatica L.) in Thuringia. Allgemeine Forst- Und Jagdzeitung 168(10): 182–190.
Kraj W., Sztorc A. (2009). Genetic structure and variability of phenological forms in the European beech (Fagus sylvatica L.). Annals of Forest Science 66(2): 203–203. http://dx.doi.org/10.1051/forest/2008085.
Kramer K., Degen B., Buschbom J., Hickler T., Thuiller W., Sykes M.T., de Winter W. (2010). Modelling exploration of the future of European beech (Fagus sylvatica L.) under climate change – range, abundance, genetic diversity and adaptive response. Forest Ecology and Management 259(11): 2213–2222. http://dx.doi.org/10.1016/j.foreco.2009.12.023.
Lander T.A., Oddou-Muratorio S., Prouillet-Leplat H., Klein E.K. (2011). Reconstruction of a beech population bottleneck using archival demographic information and Bayesian analysis of genetic data. Molecular Ecology 20: 5182–5196. http://dx.doi.org/10.1111/j.1365-294X.2011.05356.x.
Larsen A.B. (1996). Genetic structure of populations of beech (Fagus sylvatica L.) in Denmark. Scandinavian Journal of Forest Research 11(1–4): 220–232. http://dx.doi.org/10.1080/02827589609382931.
Latałowa M. (1992). Man and vegetation in the pollen diagrams from Wolin Island (NW Poland). Acta Palaeobotanica 32(1): 123–249.
Latałowa M., Ralska-Jasiewiczowa M., Miotk-Szpiganowicz G., Zachowicz J., Nalepka D. (2004). Fagus sylvatica L.– beech. In: Late Glacial and Holocene history of vegetation in Poland based on isopollen maps.W. Szafer Institute of Botany, Polish Academy of Sciences, Kraków. p. 95–104.
Lefèvre S., Wagner S., Petit R.J., De Lafontaine G. (2012). Multiplexed microsatellite markers for genetic studies of beech. Molecular Ecology Resources 12: 484–491. http://dx.doi.org/10.1111/j.1755-0998.2011.03094.x.
Leonardi S., Menozzi P. (1995). Genetic variability of Fagus sylvatica L. in Italy: the role of postglacial recolonization. Heredity 75: 35–44. http://dx.doi.org/10.1038/hdy.1995.101.
Magri D., Vegetale B., Sapienza L., Moro P.A. (2008). Patterns of post-glacial spread and the extent of glacial refugia of European beech (Fagus sylvatica). Journal of Biogeography 35: 450–463. http://dx.doi.org/10.1111/j.1365-2699.2007.01803.x.
Magri D., Vendramin G.G., Comps B., Dupanloup I., Geburek T., Gömöry D., Latałowa M., Litt T., Paule L., Roure J.M., Tantau I., van der Knaap W.O., Petit R.J., De Beaulieu J.L. (2006). A new scenario for the Quaternary history of European beech populations: palaeobotanical evidence and genetic consequences. New Phytologist 171(1): 199–221. http://dx.doi.org/10.1111/j.1469-8137.2006.01740.x.
Matuszkiewicz J.M. (2001). Zespoły leśne Polski. [Polish forest communities]. Wydawnictwo Naukowe PWN, Warszawa. 378 p.
Merzeau D., Comps B., Thiebaut B. (1994). Genetic structure of natural stands of Fagus sylvatica L. (beech). Heredity 72: 269–277. http://dx.doi.org/10.1038/hdy.1994.37.
Mitka J., Bąba W., Szczepanek K. (2014). Putative forest glacial refugia in the Western And Eastern Carpathians. Modern Phytomorphology 5: 85–92.
Netzer F., Thöm C., Celepirovic N., Ivankovic M., Alfarraj S., Dounavi A., Judy Simon J., Herschbach C., Rennenberg H. (2016). Drought effects on C, N, and P nutrition and the antioxidative system of beech seedlings depend on geographic origin. Journal of Plant Nutrition and Soil Science 179(2): 136–150. http://dx.doi.org/10.1002/jpln.201500461.
Paffetti D., Travaglini D., Buonamici A., Nocentini S., Vendramin G.G., Giannini R., Vettori C. (2012). The influence of forest management on beech (Fagus sylvatica L.) stand structure and genetic diversity. Forest Ecology and Management 284: 34–44. http://dx.doi.org/10.1016/j.foreco.2012.07.026.
Pastorelli R., Smulders M.J.M., Van’t Westende W.P.C., Vosman B., Giannini R., Vettori C., Vendramin G.G. (2003). Characterization of microsatellite markers in Fagus sylvatica L. and Fagus orientalis Lipsky. Molecular Ecology Notes 3(1): 76–78. http://dx.doi.org/10.1046/j.1471-8286.2003.00355.x.
Peakall R., Smouse P.E. (2006). GenAlEx 6: genetic analysis in Excel. Population genetic software for teaching and research. Molecular Ecology Notes 6(1): 288–295. http://dx.doi.org/10.1111/j.1471-8286.2005.01155.x.
Petit R.J., Aguinagalde I., de Beaulieu J.-L., Bittkau C., Brewer S., Cheddadi R., Ennos R., Fineschi S., Grivet D., Lascoux M., Mohanty A., Müller-Starck G., Demesure-Musch B., Palmé A., Martín J.P., Rendell S., Vendramin G.G. (2003). Glacial refugia: hotspots but not melting pots of genetic diversity. Science 300(5625): 1563–1565. http://dx.doi.org/10.1126/science.1083264.
Piotti A., Leonardi S., Buiteveld J., Geburek T., Gerber S., Kramer K., Vettori C., Vendramin G.G. (2012). Comparison of pollen gene flow among four European beech (Fagus sylvatica L.) populations characterized by different management regimes. Heredity 108(3): 322–331. http://dx.doi.org/10.1038/hdy.2011.77.
Pritchard J.K., Stephens M., Donnelly P. (2000). Inference of population structure using multilocus genotype data. Genetics 155(2): 945–59.
Rajendra K.C., Seifert S., Prinz K., Gailing O., Finkeldey R. (2014). Subtle human impacts on neutral genetic diversity and spatial patterns of genetic variation in European beech (Fagus sylvatica). Forest Ecology and Management 319: 138–149. http://dx.doi.org/10.1016/j.foreco.2014.02.003.
Ralska-Jasiewiczowa M. (1983). Isopollen maps for Poland 0–11 000 years B.P. New Phytologist 94(1): 133–175.
Ralska-Jasiewiczowa M., Nalepka D., Goslar T. (2003). Some problems of forest transformation at the transition to the oligocratic/ Homo sapiens phase of the Holocene interglacial in northern lowlands of central Europe. Vegetation History and Archaeobotany 12(4): 233–247. http://dx.doi.org/10.1007/s00334-003-0021-8.
Rose L., Leuschne C., Köckeman B., Buschmann H. (2009). Are marginal beech (Fagus sylvatica L.) provenances a source for drought tolerant ecotypes? European Journal of Forest Research 128(4): 335–343. http://dx.doi.org/10.1007/s10342-009-0268-4.
Seifert S. (2011). Variation of candidate genes related to climate change in European beech (Fagus sylvatica L .). Georg-August-Universität Göttingen.
Shanjani P., Vendramin G., Calagari M. (2011). Altitudinal genetic variations among the Fagus orientalis Lipsky populations in Iran. Iranian Journal of Biotechnology 9(1): 11–20.
Stewart J.R., Lister A.M. (2001). Cryptic northern refugia and the origins of the modern biota. Trends in Ecology & Evolution 16(11): 608–613. http://dx.doi.org/10.1016/S0169-5347(01)02338-2.
Sułkowska M. (2010). Genetic and ecotypic characterization of European beech (Fagus sylvatica L.) in Poland. Acta Silvatica et Lignaria Hungarica 6: 155–121.
Svenning J.-C., Skov F. (2007). Could the tree diversity pattern in Europe be generated by postglacial dispersal limitation? Ecology Letters 10(6): 453–460. http://dx.doi.org/10.1111/j.1461-0248.2007.01038.x.
Szafer W. (1932). The beech and the beechforest in Poland. Veröffentlichungen Des Geobotanischen Institutes Rübel in Zürich 8: 168–181.
Taberlet P., Fumagalli L., Wust-Saucy A.G., Cosson J.F. (1998). Comparative phylogeography and postglacial colonization routes in Europe. Molecular Ecology 7(4): 453–64.
Tanaka K., Tsumura Y., Nakamura T. (1999). Development and polymorphism of microsatellite markers for Fagus crenata and the closely related species, F. japonica. Theoretical and Applied Genetics 99(1–2): 11–15. http://dx.doi.org/10.1007/s001220051203.
Thuiller W., Lavorel S., Sykes M.T., Araujo M.B., Araújo M.B. (2006). Using niche-based modelling to assess the impact of climate change on tree functional diversity in Europe. Diversity and Distributions 12(1): 49–60. http://dx.doi.org/10.1111/j.1366-9516.2006.00216.x.
Tzedakis P.C., Emerson B.C., Hewitt G.M., Sa F., Laguna L., Islands C. (2013). Cryptic or mystic? Glacial tree refugia in northern Europe. Trends in Ecology & Evolution 28(12): 696–704. http://dx.doi.org/10.1016/j.tree.2013.09.001.
Van Oosterhout C., Van Heuven M.K., Brakefield P.M. (2004). On the neutrality of molecular genetic markers: pedigree analysis of genetic variation in fragmented populations. Molecular Ecology 13(5): 1025–1034. http://dx.doi.org/10.1111/j.1365-294X.2004.02114.x.
Van Oosterhout C., Weetman D., Hutchinson W.F. (2006). Estimation and adjustment of microsatellite null alleles in nonequilibrium populations. Molecular Ecology Notes 6(1): 255–256. http://dx.doi.org/10.1111/j.1471-8286.2005.01082.x.
Vornam B., Decarli N., Gailing O. (2004). Spatial distribution of genetic variation in a natural beech stand (Fagus sylvatica L.) based on microsatellite markers. Conservation Genetics 5(4): 561–570. http://dx.doi.org/10.1023/B:COGE.0000041025.82917.ac.
Weir B.S., Cockerham C.C. (1984). Estimating F-statistics for the analysis of population structure. Evolution 38(6): 1358. http://dx.doi.org/10.2307/2408641.
Widmer A., Lexer C. (2001). Glacial refugia: sanctuaries for allelic richness, but not for gene diversity. Trends in Ecology & Evolution 16(6): 267–269. http://dx.doi.org/10.1016/S0169-5347(01)02163-2.
Williams A.P., Allen C.D., Macalady A.K., Griffin D., Woodhouse C.A., Meko D.M., Swetnam T.W., Rauscher S.A., Seager R., Grissino-Mayer H.D., Dean J.S., Cook E.R., Gangodagamage C., Cai M., McDowell N.G. (2012). Temperature as a potent driver of regional forest drought stress and tree mortality. Nature Climate Change 3(3): 292–297. http://dx.doi.org/10.1038/nclimate1693.
Willis K., van Andel T. (2004). Trees or no trees? The environments of central and eastern Europe during the last glaciation. Quaternary Science Reviews 23(23–24): 2369–2387. http://dx.doi.org/10.1016/j.quascirev.2004.06.002.
Willis K.J., Rudner E., Sümegi P. (2000). The full-glacial forests of central and southeastern Europe. Quaternary Research 53(2): 203–213. http://dx.doi.org/10.1006/qres.1999.2119.
Total of 82 references.