Fei-Fei Zhai,
Jun-Xiang Liu,
Zhen-Jian Li,
Jin-Mei Mao,
Yong-Qiang Qian,
Lei Han,
Zhen-Yuan Sun 
Assessing genetic diversity and population structure of Salix viminalis across Ergun and West Liao basin
Zhai F.-F., Liu J.-X., Li Z.-J., Mao J.-M., Qian Y.-Q., Han L., Sun Z.-Y. (2017). Assessing genetic diversity and population structure of Salix viminalis across Ergun and West Liao basin. Silva Fennica vol. 51 no. 3 article id 7001. https://doi.org/10.14214/sf.7001
Highlights
- High genetic diversity and moderate population differentiation were detected in twelve Salix viminalis populations
- Populations from Ergun basin presented higher genetic diversity than populations from West Liao basin
- Populations with short distances presented low differentiation.
Abstract
Salix viminalis L. is an important shrub that has potential for use as a bioenergy crop, for phytoremediation of heavy metal contaminated soil and sewage sludge treatment. It is mainly distributed in the northeast of China, but the species has not yet been used a resource here. We examined the genetic diversity and population structure of populations from the Ergun basin and West Liao basin using 20 microsatellite markers. A high level of genetic diversity (Na = 16.45, He = 0.742) was detected for S. viminalis, and populations from the Ergun basin exhibited higher genetic diversity and private alleles numbers than the West Liao basin. The 12 populations could be divided into two clusters by both Bayesian analysis and UPGMA clustering which were consistent with the populations derived from the two basins. Moderate population differentiation (FST = 0.076) was shown in S. viminalis, and AMOVA analysis confirmed that most of the genetic variation (86.13%) was attributed to individual differences within populations, while 11.49% was attributed to differences between basins and 2.38% to differences within each basin. Significant correlations of FST/(1–FST) with log (geographic distance) among 12 populations (r = 0.634, p < 0.00) and 10 populations within the Ergun basin (r = 0.482, p = 0.0002) indicated that geographical distance was the principal factor influencing genetic structure. As most of genetic variation exist within populations, so protection measures should be focused on populations with higher genetic diversity and unique alleles, such as Tuli, Mordaga downstream, Zhadun1 and Genhe.
Keywords
genetic variation;
microsatellites;
geographic barrier;
basket willow;
common osier
- Zhai, State Key Laboratory of Tree Genetics and Breeding; Research Institute of Forestry, Chinese Academy of Forestry; Key Laboratory of Tree Breeding and Cultivation, State Forestry Administration, Haidian District, Beijing, 10091, PR China; School of Architectural and Artistic Design, Henan Polytechnic University, Century Avenue, Jiaozuo, Henan, 454000, PR China E-mail lkyzff@163.com
- Liu, State Key Laboratory of Tree Genetics and Breeding; Research Institute of Forestry, Chinese Academy of Forestry; Key Laboratory of Tree Breeding and Cultivation, State Forestry Administration, Haidian District, Beijing, 10091, PR China E-mail liu-jx295@163.com
- Li, State Key Laboratory of Tree Genetics and Breeding; Research Institute of Forestry, Chinese Academy of Forestry; Key Laboratory of Tree Breeding and Cultivation, State Forestry Administration, Haidian District, Beijing, 10091, PR China E-mail zhenjianli@163.com
- Mao, State Key Laboratory of Tree Genetics and Breeding; Research Institute of Forestry, Chinese Academy of Forestry; Key Laboratory of Tree Breeding and Cultivation, State Forestry Administration, Haidian District, Beijing, 10091, PR China; Research Institute of Economic Forest, Xinjiang Academy of Forestry, Anjunanlu, Urumqi, Xinjiang, PR China E-mail 350512173@qq.com
- Qian, State Key Laboratory of Tree Genetics and Breeding; Research Institute of Forestry, Chinese Academy of Forestry; Key Laboratory of Tree Breeding and Cultivation, State Forestry Administration, Haidian District, Beijing, 10091, PR China E-mail qianyq@caf.ac.cn
- Han, State Key Laboratory of Tree Genetics and Breeding; Research Institute of Forestry, Chinese Academy of Forestry; Key Laboratory of Tree Breeding and Cultivation, State Forestry Administration, Haidian District, Beijing, 10091, PR China E-mail hdd@caf.ac.cn
-
Sun,
State Key Laboratory of Tree Genetics and Breeding; Research Institute of Forestry, Chinese Academy of Forestry; Key Laboratory of Tree Breeding and Cultivation, State Forestry Administration, Haidian District, Beijing, 10091, PR China
E-mail
lkyszy@126.com
Received 16 March 2017 Accepted 30 May 2017 Published 16 June 2017
Views 162541
Available at https://doi.org/10.14214/sf.7001 | Download PDF

Supplementary Files
1 Introduction
The genus Salix (willow) have been considered one important taxonomic entity in the world because of its extensive use, such as riverbank stabilization (Watson et al. 1997), medical uses (Mahdi et al. 2010) and basket production (Grönroos et al. 1989). The genus comprises around 520 species worldwide which show considerable variation in plant size, growth habit and morphological characteristics, ranging from small shrubs to large trees (Fang et al. 1987; Newsholme 1992). Salix spp. occupy diverse ecological niches, and most of them are native to the northern hemisphere (Argus 1997). Asia is considered to be the original center of genus Salix and about 275 species exist in China, of which 189 are endemics (Fang et al. 1987).
Salix viminalis L. belongs to family Salicaceae. It is a diploid species (2n = 2x = 38) with an outcrossing breeding system that combines insect and wind pollination (Rönnberg-Wästljung 2001; Hanley et al. 2002). S. viminalis is a pioneer tree species which is mainly found in riparian environments, and it can be propagated both sexually and vegetatively (Flora of China Editorial Committee 1994). The willow is prized for its economic value as a promising woody bioenergy crop due to perennial habit, easy vegetative propagation, fast growth rate and good coppicing response (Keoleian and Volk 2005; Trybush et al. 2012). In recent years, S. viminalis also be used for sewage sludge treatment (Kocik et al. 2007) and phytoremediation of heavy metal contaminated soils (Klang-Westin and Perttu 2002; Mleczek et al. 2010). S. viminalis is a diecious shrub native to China, which is mainly distributed across the northeast, such as Heilongjiang, Jilin, Liaoning and eastern Inner Mongolia provinces (Flora of China Editorial Committee 1994). So far, S. viminalis has been underutilized and the overwhelming majority of this specie is still living in the wild in China. Moreover, with the increase of human activities, the survival of S. viminalis has been strongly impacted. Assessment of the genetic diversity and population structure of natural S. viminalis is essential to conduct species’ conservation and initiate domestication process.
Over the last decades, a number of molecular marker technologies have appeared and been proved to be valuable tools to evaluate genetic diversity within and between populations (Kadri et al. 2009). Among various molecular markers, microsatellite or simple sequence repeat (SSR) makers are well known for multiallelic nature, codominant inheritance, high information content, and simple detection by PCR amplifications (Wang et al. 2012). Many SSR markers have been developed in Salix genus, and it is possible to use these markers in related species within the same genus because of the transferability of the markers (Lian et al. 2002; Barker et al. 2003; Du et al. 2012). There have been studies conducted to assess the genetic diversity of S. viminalis (Trybush et al. 2012; Lascoux et al. 1996; Przyborowski and Sulima 2010). However, the number of individuals within populations was limited, and current researches are confined to central Europe.
In this study, we collected cuttings of 330 S. viminalis individuals from 12 natural populations across Ergun basin and West Liao basin in China, and all samples were analyzed using 20 SSR markers obtained from literatures. The specific aims of this study were: (1) to evaluate genetic variation within and between populations (2) to examine whether the genetic diversity of populations from West Liao basin was the same as that of populations from Ergun basin and (3) assess genetic differentiation of populations with short distances.
2 Material and methods
2.1 Plant materials and DNA extraction
Cuttings of 330 S. viminalis individuals were collected from 12 populations along 9 rivers across two basins (Table1; Fig.1). The river Mordaga, Genhe, Tuli, Kuduer, Zhadun, Yimin and Huihe belong to Ergun basin, while river Dahaiqing and Heili belong to West Liao basin. Moreover, three populations were collected along Zhadun river to study genetic differentiation of populations with short distance. For each population, 17–30 individuals were randomly sampled. In order to avoid resampling clones, the individuals within population should be at least 50 m apart (except at YM, HH and HL sites where willows are no more than 50). As soon as the cuttings were transported to laboratory, they were planted in pot at once. One month later, fresh leaves were sampled and kept frozen at –80 °C in laboratory for later use. Total DNA was extracted from about 150 mg fresh leaf tissues according to the modified cetyltrimethyl ammonium bromide (CTAB) protocol described by Doyle and Doyle (1987). The DNA concentration was checked with a spectrophotometer (NAS-99, ACTGene Company) and the DNA quality was verified on a 1.2% agarose gel electrophoresis. DNA samples were stored at –18 °C.
| Table 1. General information for 12 Salix viminalis populations across two basins in China. | |||||
| Population | River | Basin | Latitude, Longitude | Altitude (m a.s.l.) | Samples |
| MDG1 | Mordaga upstream | Ergun | 51°16´N, 120°44´E | 673 | 30 |
| MDG2 | Mordaga downstream | Ergun | 51°25´N, 120°00´E | 572 | 30 |
| GH | Genhe | Ergun | 50°46´N, 121°30´E | 706 | 30 |
| TL | Tuli | Ergun | 50°29´N, 121°41´E | 734 | 28 |
| KDE | Kuduer | Ergun | 50°02´N, 121°40´E | 848 | 27 |
| ZD1 | Zhadun | Ergun | 49°20´N, 120°40´E | 649 | 30 |
| ZD2 | Zhadun | Ergun | 49°20´N, 120°41´E | 650 | 30 |
| ZD3 | Zhadun | Ergun | 49°19´N, 120°40´E | 650 | 30 |
| YM | Yimin tributary | Ergun | 48°42´N, 119°47´E | 662 | 18 |
| HH | Huihe | Ergun | 48°04´N, 119°38´E | 772 | 17 |
| DHQ | Dahaiqing | West Liao | 44°14´N, 118°20´E | 1162 | 30 |
| HL | Heili | West Liao | 41°21´N, 118°27´E | 1049 | 30 |
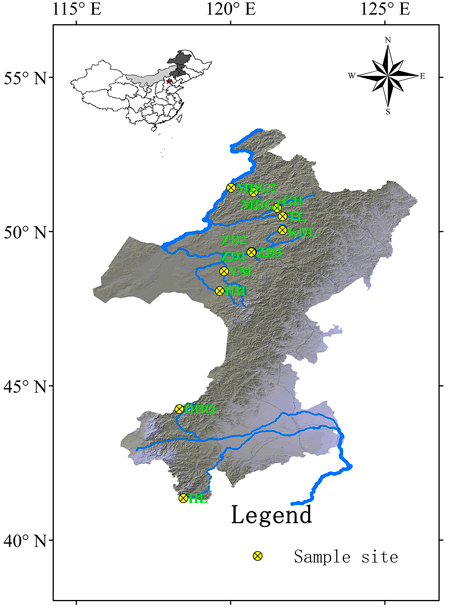
Fig. 1. Geographic locations of 12 Salix viminalis populations across two basins in China. [Note: Abbreviations of populations are explained in Table 1].
2.2 PCR amplification and genotyping
A total of 70 microsatellite markers of Salix were selected from public literatures (Lian et al. 2001; Barker et al. 2003; Stamati et al. 2003; Hoshikawa et al. 2009). They were used to detect inter-specific variation across six S. viminalis individuals by polyacrylamide gel electrophoresis. Based on the clarity and reproducibility of the band patterns, twenty microsatellite markers revealed high inter-individual variation and then were amplified to all samples. Amplification reactions were performed in a 15 μl volume containing 6.0 µl of 2.5 × PCR buffer (KAPA Taq HotStart PCR Kits, KAPA Company), 1.0 µl of each of the forward and reverse primers (5 µM), 0.1µl of Taq polymerase (5U µl–1), 1.0 µl of template DNA, and water to 15 µl. The thermal cycling was performed using GeneAmp 9600 PCR system (Applied Biosystems) as follows: 10 min at 95 °C; then 30 cycles consisting of 30 s at 95 °C, 30 s at 58 °C, 30 s at 70 °C, followed by a final extension for 7 min at 72 °C. After amplification, 1 µl of PCR products were added to 0.5 µl of ROX-500 size standard (Beijing Microread Gene Technology Co., Ltd) and 8.5 µl of Hi-Di formamide (Applied Biosystems) in 96 well-plates, and denatured at 95 °C for 3 min. Then the products were separated by capillary electrophoresis on 3730XL DNA analyzer (Applied Biosystems). Data were scored using the GeneMapper v 4.0 (Applied Biosystems).
2.3 Data analysis
The intra- and inter-population genetic statistics were measured using POPGENE v. 1.32 (Yeh et al. 1997) and GenAlex 6.5 (Peakall and Smouse 2012), including number of observed alleles (Na), number of effective alleles (Ne), observed heterozygosity (Ho), expected heterozygosity (He), number of private alleles (Np) and F-statistics (FIS, FIT, FST). Besides, statistical tests were performed on SPSS 18.0 (SPSS Inc., Chicago, IL, USA) with one-way analysis of variance to reveal the difference of two basins.
Without prior classification information, population structure can be analyzed by Bayesian clustering algorithm in software STRUCTURE v 2.3.4 (Falush et al. 2003; Pritchard et al. 2000). Fifteen independent runs were performed for each value of K ranging from 1 to 12 with a burn-in of 50 000 iterations followed by 100 000 iterations. The most appropriate K value could be determined by mathematical model described by Pritchard et al. (2000) and Evanno et al. (2005). Genetic relationships among populations were performed using 10 000 bootstrap replicates from PowerMarker v. 3.25 (Liu and Muse 2005) based on Shriver distance (Shriver et al. 1995) and a UPGMA tree was constructed with Consense of the software package Phylip v. 3.6 (Felsenstein 2004). Furthermore, in order to investigate the correlation between genetic and geographic distance between all pairs of populations, Isolation By Distance (IBD) analysis based on a mantel test with 30 000 matrix randomizations was carried out using IBDWS (isolation by distance web service) (Jensen et al. 2005). The software MEGA 6.0 (Tamura et al. 2013) was applied to view and edit the tree. Analysis of molecular variance (AMOVA) was performed on GenAlex to estimate the genetic variation within and among populations. Pairwise values of genetic differentiation (FST) were calculated by the AMOVA routine in GenAlex, and permutation procedures (9999 replicates) were used to test the significance of the differentiation between pairs of populations. In addition, the program Barrier version 2.2 was used to identify potential physical barriers (Manni et al. 2004).
3 Results
3.1 Polymorphism of 20 simple sequence repeat (SSR) markers
A total of 329 alleles at 20 SSR markers were identified across 330 samples and all of the markers showed moderate to high level of polymorphism. The number of observed alleles (Na) ranged from 3 at SB1148 to 49 at gSIMCT052 with an average of 16.45, while the number of effective alleles (Ne) ranged from 1.595 at Shuk058 to 13.47 at gSIMCT052 with an average of 5.384. Mean observed heterozygosity (Ho) and expected heterozygosity (He) were 0.636 and 0.742, respectively; Ho was less than He at 18 loci except SB1172 and SB1148 which were confirmed by FIS and FIT, indicating a deficiency of heterozygote at these 18 loci (Supplementary file S1).
3.2 Genetic diversity of Salix viminalis populations
The mean Na and Ne per locus and per population were 8.313 and 4.387, respectively. The maximum values of Na (10.50) and Ne (5.549) presented in TL and KDE separately, while the minimum values of them (Na = 3.850, Ne = 2.187) appeared in HL. Observed heterozygosity (Ho) ranged from 0.490 (HL) to 0.728 (KDE), while expected heterozygosity (He) ranged from 0.473 (HL) to 0.757 (KDE) (Table 2). For each population, Ho was less than He except HL where an excess of heterozygote were observed. A total of 56 private alleles were found in 12 populations ranging from 0(ZD2) to 14 (TL), with an average value of 4.67 (Table 2). Moreover, the genetic diversity between the two basins showed highly significant differences by one-way analysis of variance (Suppl. file S2).
| Table 2. Genetic diversity of 12 populations in Salix viminalis based on 20 SSR markers. | ||||||
| Population | Na | Ne | Ho | He | FIS | Np |
| MDG1 | 8.600 | 4.701 | 0.671 | 0.737 | 0.073 | 3 |
| MDG2 | 9.150 | 4.743 | 0.637 | 0.719 | 0.098 | 8 |
| GH | 9.600 | 5.176 | 0.657 | 0.734 | 0.083 | 6 |
| TL | 10.50 | 5.524 | 0.650 | 0.754 | 0.120 | 14 |
| KDE | 9.100 | 5.549 | 0.728 | 0.757 | 0.020 | 7 |
| ZD1 | 9.200 | 4.275 | 0.667 | 0.705 | 0.037 | 8 |
| ZD2 | 8.950 | 4.596 | 0.650 | 0.726 | 0.079 | 0 |
| ZD3 | 9.400 | 4.709 | 0.655 | 0.748 | 0.104 | 4 |
| YM | 8.200 | 4.220 | 0.658 | 0.712 | 0.043 | 2 |
| HH | 7.050 | 4.143 | 0.653 | 0.718 | 0.072 | 1 |
| DHQ | 6.150 | 2.825 | 0.542 | 0.587 | 0.064 | 1 |
| HL | 3.850 | 2.187 | 0.490 | 0.473 | –0.059 | 2 |
| Mean | 8.313 | 4.387 | 0.638 | 0.698 | 0.062 | 4.670 |
| Abbreviations of populations are explained in Table 1. Na = Number of observed alleles; Ne = Number of effective alleles; Ho = Observed heterozygosity; He = Expected heterozygosity; FIS = Inbreeding coefficient among individuals within subpopulation; Np = Number of private alleles. | ||||||
3.3 Genetic structure of Salix viminalis populations
According to the mathematical model described by Pritchard et al. (2000), the most optimal K value was to be 3 where the log-likelihood value emerged an inflection point (Suppl. file S3). However, the statistical model suggested by Evanno et al. (2005) showed a clear peak at value of K = 2 (Suppl. file S3). The clustering results of 12 populations are shown in Fig. 2. When K = 2, the 12 populations could be divided into two clusters: the first cluster comprised individuals from HL and DHQ of West Liao basin, while the second cluster comprised individuals from Ergun basin. When K = 3, the 12 populations could be divided in to three clusters: the individuals of West Liao basin were still classified into the same cluster, while individuals of Ergun basin could be divided into two clusters equaling that the individuals from Ergun basin were with two substructures: populations of GH, KDE, MDG1, MDG2 and TL constituted one subcluster (S1), and HH, YM, ZD1, ZD2 and ZD3 constituted the other subcluster (S2). The results of Bayesian clustering were confirmed by UPGMA with strong bootstrap supporting for Ergun cluster and West Liao cluster, although subclusters in Ergun cluster were not obvious (Fig. 3). This was further supported by Mantel test with 30 000 permutations, revealing significant correlation between FST/(1–FST) and log (geographic distance) among 12 populations (y = 0.153x – 0.261, r = 0.634, p < 0.00, Fig. 4A). In addition, significant correlations of genetic distance and geographic distance was also observed in Ergun basin (y = 0.031x – 0.038, r = 0.482, p = 0.0002, Fig. 4B).
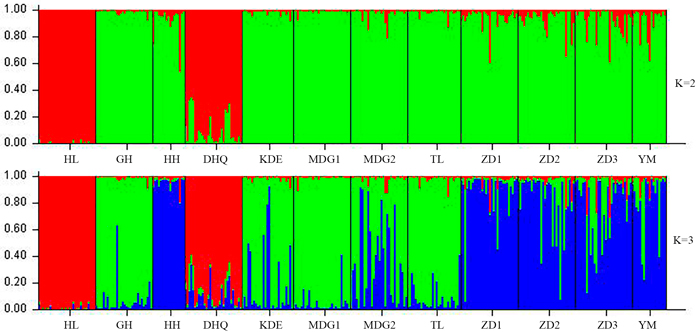
Fig. 2. Estimated population structure of 330 individuals in Salix viminalis with K = 2 and 3. [Note: Abbreviations of populations are explained in Table 1].
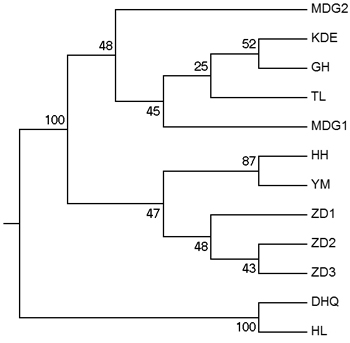
Fig. 3. UPGMA tree of 12 populations based on Shriver distance using 10 000 bootstrap replicates. [Note: Abbreviations of populations are explained in Table 1].
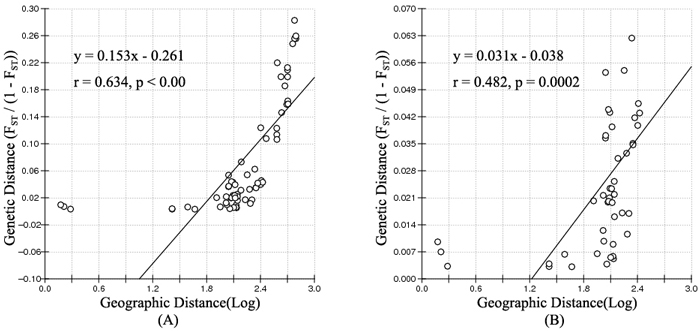
Fig. 4. Isolation By Distance (IBD) analysis between FST/(1–FST) and log (geographic distance) among 12 Salix viminalis populations (A) and among 10 populations in Ergun basin (B).
3.4 Genetic differentiation and genetic variance of Salix viminalis populations
For the 12 populations of S. viminalis, FST of different loci ranged from 0.031 to 0.141 with an average of 0.076 (Suppl. file S1), while pairwise FST values ranged from 0.003 to 0.220 (Suppl. file S4). Highly significant differentiation between populations of two basins was revealed by pairwise FST values, and the differentiation of the two subclusters of Ergun basin was also highly significant. However, there was no statistically significant differentiation between HH and YM, ZD2 and ZD3, and among KDE, TL, GH and MDG1 (Suppl. file S4).
AMOVA analysis revealed the populations were structured and moderately differentiated. As expected, the largest proportion of the total molecular variation (93.37%) was attributed to differences within populations and 6.63% was among populations. If taking into account the clustering results by Bayesian and Neighbor-Joining clustering, then most of the total genetic variation (86.13%) could be due to individual differences within populations while 11.49% was ascribed to difference of two basins and 2.38% existed in populations within each basin. For populations of Ergun basin, the variation between subclusters (2.33%) was larger than that between populations within subclusters (0.88%), but 96.80% of variation was still associated with individual differences within populations (Table 3).
| Table 3. Analysis of Molecular Variance (AMOVA) of 12 Salix viminalis populations. | |||||
| Source of variation | Degrees of freedom | Sum of squares | Mean Squares | Estimated variance | Explained variance (%) |
| Total populations | |||||
| Between populations | 11 | 382.05 | 34.73 | 0.795 | 6.63 |
| Within populations | 648 | 4506.32 | 13.93 | 6.97 | 93.37 |
| Two clusters (two basins) | |||||
| Between clusters | 1 | 201.40 | 201.40 | 0.93 | 11.49 |
| Between populations within clusters | 10 | 180.65 | 18.07 | 0.19 | 2.38 |
| Within populations | 648 | 4506.33 | 13.93 | 6.97 | 86.13 |
| Two subclusters of Ergun basin | |||||
| Between subclusters | 1 | 59.07 | 59.07 | 0.176 | 2.33 |
| Between populations within subclusters | 8 | 92.88 | 11.61 | 0.066 | 0.88 |
| Within populations | 530 | 3880.71 | 14.67 | 7.34 | 96.80 |
Results revealed by Barrier software was in accord with the analyses of genetic structure. When the number of barrier was set as only one, a putative barrier (Da Hinggan Mountains) which separated the twelve populations into two groups was detected. Moreover, there were other two barriers existing in two basins respectively when the number of barrier was set to three. One isolated HL from DHQ and the other isolated HH, YM, ZD1, ZD2 and ZD3 from MDG1, MDG2, GH, TL and KDE (Fig. 5).
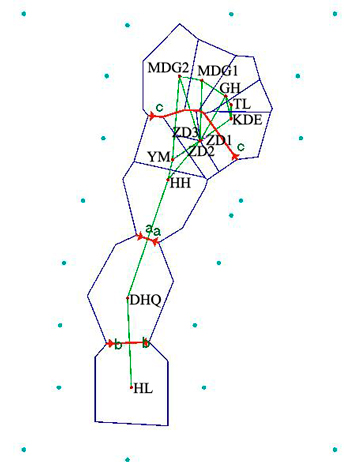
Fig. 5. Genetic barrier of 12 Salix viminalis populations based on Barrier version 2.2. [Note: Abbreviations of populations are explained in Table 1].
4 Discussion
4.1 High polymorphism of simple sequence repeat (SSR) markers and high genetic diversity of Salix viminalis
The application of SSR markers to estimate the level of genetic diversity has been shown to be appropriate in many studies (Liu et al. 2012; Trybush et al. 2012; Talve et al. 2014). In this study, a moderate to high level of polymorphism for the 20 SSR markers indicates that they are effective to reveal genetic variation of S. viminalis. An average number of 16.45 observed alleles (Na) and 5.38 effective alleles (Ne) per locus within 330 individuals was higher than studies in Salix like S. caprea (Na = 10.00, Ne = 3.90) (Perdereau et al. 2014), and S. viminalis in Czech Republic (Na = 13.46, Ne = 3.73) (Trybush et al. 2012). Relatively high value of observed heterozygosity (Ho) (0.64) and He (0.74) are comparable to the diversity of Populus nigra (Ho = 0.70, He = 0.73) (Rathmacher et al. 2010) and S. viminalis (Ho = 0.55, He = 0.56) (Berlin et al. 2014). The high genetic diversity of S. viminalis is probably associated with its biological characters including woody habit, dioecism, wind or insect pollination, and wind seed dispersal (Hamrick et al. 1992; Karp et al. 2011; Teixeira et al. 2014). Moreover, heterozygosis deficit is observed within all populations except HL. The deficit of heterozygosity is very common in the Salicaceae family, such as S. lapponum and S. lanata (Stamati et al. 2007), S. daphnoides (Sochor et al. 2013), P. alba (Castiglione et al. 2010) and P. davidiana (Lee et al. 2011). Limited population size, extensive clonality or biased sex ratio and subsequent inbreeding are usually the reasons of deficit (Mandel et al. 2001; Sochor et al. 2013).
The SSR data demonstrated that the populations from Ergun basin exhibited significantly more genetic diverse than that from West Liao basin. The two populations of Ergun basin were either small or isolated, genetic drift are likely to take place. A small number of private alleles were also detected in the two populations. The genetic drift, small efficient individuals and inbreeding would lead the genetic diversity to decrease in Ergun basin. Chen et al. (2008) found that the genetic variance might be lost under unfavorable environment due to an increase in vegetative reproduction and a decrease in resource-demanding sexuality. The unfavorable environment in West Liao basin with arid spring and hot summer might also lead to declining genetic diversity. In this study, lower genetic diversity and together with less number of private alleles for populations from West Liao basin might also reflect changes of geographic distribution caused by past climate changes and glaciation followed by an expansion of S. viminalis distribution towards lower latitudes (Kim et al. 2011; Mehes et al. 2009). Less private alleles found in YM, HH and ZD2 of Ergun basin might be due to the disturbance of human activities, leading to rapid reduction in individuals.
4.2 Geographical patterns of Salix viminalis
The evolutionary potential and ability to cope with adverse environment of a species not only depends on intraspecific genetic variation, but also determined by genetic structure of population (Millar and Libby 1991). Both of Bayesian analysis and UPGMA clustering suggested that the 12 populations could be classified to two different genetic clusters which generally agreed with populations deriving from two basins. The populations of Ergun basin were further subdivided into two subclusters. It has been reported that physical barriers, such as mountains, rivers and deserts often play an important role in the population structure (Lamborot and Eaton 1997; Yuan et al. 2012; Wang et al. 2015). The Barrier result showed that the most significant geographical barrier was between the two basins. Da Hinggan Mountains could act as a key barrier to impede gene exchange among populations of two basins. In addition, the two suclesters of Ergun basin are located on the east and west side of Da Hinggan Mountains respectively, so Da Hinggan Mountains was also a barrier for populations inside the Ergun basin.
The overall genetic differentiation based on FST (0.076 on average) showed that 7.60% of the total genetic variation was attributed to inter-population differentiation and 92.84% to individual differentiation within populations, suggesting moderate differentiation of S. viminalis according to the criterion suggested by Wright (1978). AMOVA results also showed a moderate differentiation that 6.63% genetic variance was among populations, which was mainly due to the contrast between two basins (Table 3). Our result was consistent with previous studies that the differentiation was low to moderate for species of Saliaceae (Imbert and Lefèvre 2003; Smulders et al. 2008; Lin et al. 2009; Callahan et al. 2013). However, the value of FST in our study is higher than that for natural populations of S. viminalis in Czech Republic (FST = 0.05) (Trybush et al. 2012), which might be due to that the 12 populations in our study are distributed in two basins with long distance, and they are separated and isolated by Da Hinggan Mountains between them.
Isolation By Distance (IBD) is based on the theory that when the populations are in equilibrium between gene flow and gene drift, a positive relationship should be detected between genetic distance and geographic distance. The mantel test showed a significant positive correlation of
FST/(1–FST) with log (geographic distance) among all 12 populations and among 10 populations in Ergun basin. It means that the genetic structure of S. viminalis is affected by geographic isolation and this is the results of adapting environment for a long time. Conversely, no correlation was found between FST/(1–FST) and geographic distance of 6 S. viminalis populations in Czech republic (r = 0.316, p = 0.262) (Trybush et al. 2012). The inconsistent results might be due to that the populations in our study have little disturbance by human activities and the sampling range is relatively larger. Though YM, HH and ZD2 have been disturbed to some extent, it mainly occurs in recent years.
Populations of KDE, TL, GH and MDG1 were not statistically differentiated. The exchange of material that local residents collected twigs of S. viminalis for firewood or weaving might explain this result. Populations of ZD, YM and HH are located in plain area and the three rivers join into Hailar river and flood events could promote widespread regeneration across a floodplain (Hughes and Rood 2003). For species with remarkable capacity to flooding, they can even grow vigorously in flooded conditions (Bailey-Serres and Voesenek 2008). The flood in Hailar river might promote the gene exchange of ZD, YM and HH populations, so no differentiation was found. However, the flood has little effect on the differentiation of other populations of the same basin because they were located in mountain area. The differentiation is usually assumed very weak in Salicaceae, but the gene flow was restricted within 1–3 km for P. nigra (Imbert and Lefèvre 2003). In our study, differentiation of the three populations with short distance (1.5~1.9 km) from Zhadun river was very low, which was probably related to the outbreeding system of S. viminalis. The three sites along Zhadun river bank are not separated by barriers such as mountain, so the gene exchange could take place within above distances widely.
5 Conclusion
This study applied 20 SSR markers to examine the genetic diversity and genetic structure in natural populations of S. viminalis across Ergun and West Liao basins, China. Genotyping with microsatellites markers indicates that the S. viminalis are characterized by a high level genetic diversity and moderate population differentiation. The results support the separation of 12 populations into two differentiated genetic clusters which is in accordance with geographical distribution of S. viminalis populations across two basins, and Da Hinggan Mountains acts as a geographical barrier for this structure. Moreover, populations from Ergun basin exhibit significantly higher genetic diversity and higher number of private alleles than that from West Liao basin. In addition, the differentiation of the three populations with short distance (1.5~1.9 km) from Zhadun river was very low. Due to most of genetic variation exist within populations, so protection measures should be focused on populations, especially populations with higher diversity and unique alleles, such as TL, MDG2, ZD1 and GH. Meanwhile, the aforementioned populations are important reservoirs of genetic variation for special germplasm resources, thus we can make use of these potential germplasm in genetic improvement for selection of genotypes with rapid growth rate, high biomass, strong adaptability and resistance.
Acknowledgements
The work presented here was supported by the Special Fund for Forest Scientific Research in the Public Welfare (201304115), Fundamental Research Funds for the Central Non-profit Research Institution of CAF (CAFYBB2016ZX001-3) and Doctoral Scientific Fund Project of Henan Polytechnic University (B2017-40). We would like to express our sincere gratitude to Zhi-gang Yan and Rong-kui Li (Research Institute of Forestry Science and Technology, Da Hinggan Mountains in Inner Mongolia) for collecting samples across Ergun basin, and Yu-jun Lin (Forestry Bureau of Songshan District, Chifeng City) for sampling across West Liao basin. We sincerely thank Nan Jiang of Chinese Academy of Forestry for her help in drawing picture.
References
Argus G.W. (1997). Infrageneric classification of Salix (Salicaceae) in the new world. Systematic Botany Monographs 52: 1–121. https://doi.org/10.2307/25096638.
Bailey-Serres J., Voesenek L. (2008). Flooding stress: acclimations and genetic diversity. Annual Review of Plant Biology 59: 313–339. https://doi.org/10.1146/annurev.arplant.59.032607.092752.
Barker J.H.A., Pahlich A., Trybush S., Edwards K.J., Karp A. (2003). Microsatellite markers for diverse Salix species. Molecular Ecology Notes 3: 4–6. https://doi.org/10.1046/j.1471-8286.2003.00332.x.
Berlin S., Trybush S.O., Fogelqvist J., Gyllenstrand N., Hallingbaeck H.R., Åhman I. (2014). Genetic diversity, population structure and phenotypic variation in European Salix viminalis L. (Salicaceae). Tree Genetics & Genomes 10(6): 1595–1610. https://doi.org/10.1007/s11295-014-0782-5.
Callahan C.M., Rowe C.A., Ryel R.J., Shaw J.D., Madritch M.D., Mock K.E. (2013). Continental-scale assessment of genetic diversity and population structure in quaking aspen (Populus tremuloides). Journal of Biogeography 40: 1780–1791. https://doi.org/10.1111/jbi.12115.
Castiglione S., Cicatelli A., Lupi R., Patrignani G., Fossati T., Brundu G. (2010). Genetic structure and introgression in riparian populations of Populus alba L. Plant Biosystems 144(3): 656–668. https://doi.org/10.1080/11263504.2010.496188.
Chen G., Wang Y., Zhao C., Korpelainen H., Li C. (2008). Genetic diversity of Hippophae rhamnoides populations at varying altitudes in the Wolong Natural Reserve of China as revealed by ISSR markers. Silvae Genetica 57: 29–36.
Doyle J.J., Doyle J.L. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19: 11–15.
Du Q., Wang B., Wei Z., Zhang D., Li B. (2012). Genetic diversity and population structure of Chinese white poplar (Populus tomentosa) revealed by SSR markers. Journal of Heredity 103(6): 853–862. https://doi.org/10.1093/jhered/ess061.
Evanno G., Regnaut S., Goudet J. (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Molecular Ecology 14: 2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x.
Falush D., Stephens M., Pritchard J.K. (2003). Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164(4): 1567–1587. https://doi.org/10.3410/f.1015548.197423.
Fang Z.F. (1987). On the distribution and origin of Salix in the world. Chinese Acta Phytotax Sinica 25: 307–313.
Felsenstein J. (2004). PHYLIP (Phylogeny Inference Package) version 3.6. Distributed by the author. http://evolution.gs.washington.edu/phylip.html.
Flora of China Editorial Committee (1994). Flora of China. http://www.efloras.org.
Grönroos L., Von Arnold S., Eriksson T. (1989). Callus production and somatic embryogenesis from floral explants of basket willow (Salix viminalis L.). Journal of Plant Physiology 134(5): 558–566. https://doi.org/10.1016/S0176-1617(89)80147-6.
Hamrick J.L., Godt M.J.W., Sherman-Broyles S.L. (1992). Factors influencing levels of genetic diversity in woody plant species. New Forests 6: 95–124. https://doi.org/10.1007/978-94-011-2815-5_7.
Hanley S., Barker J., Van Ooijen J., Aldam C., Harris S., Åhman I., Larsson S., Karp A. (2002). A genetic linkage map of willow (Salix viminalis) based on AFLP and microsatellite markers. Theoretical and Applied Genetics 105(6): 1087–1096. https://doi.org/10.1007/s00122-002-0979-0.
Hoshikawa T., Kikuchi S., Nagamitsu T., Tomaru N. (2009). Eighteen microsatellite loci in Salix arbutifolia (Salicaceae) and cross-species amplification in Salix and Populus species. Molecular Ecology Resources 9: 1202–1205. https://doi.org/10.1111/j.1755-0998.2009.02607.x.
Hughes F.M.R., Rood S.B. (2003). Allocation of river flows for restoration of floodplain forest ecosystems: a review of approaches and their applicability in Europe. Environmental Management 32(1): 12–33. https://doi.org/10.1007/s00267-003-2834-8.
Imbert E., Lefèvre F. (2003). Dispersal and gene flow of Populus nigra (Salicaceae) along a dynamic river system. Journal of Ecology 91: 447–456. https://doi.org/10.1046/j.1365-2745.2003.00772.x.
Jensen J.L., Bohonak A.J., Kelley S.T. (2005). Isolation by distance, web service. BMC Genetics 6: 13. https://doi.org/10.1186/1471-2156-6-13.
Kadri K., Abdellawi R., Cheikh-Mhamed H. (2009). Genetic diversity in barley genetic diversity in local Tunisian barley based on RAPD and SSR analysis. Biological Diversity and Conservation 2: 27–35.
Karp A., Hanley S.J., Trybush S.O., Macalpine W., Pei M., Shield I. (2011). Genetic improvement of willow for bioenergy and biofuelsfree access. Journal of Integrative Plant Biology 53: 151–165. https://doi.org/10.1111/j.1744-7909.2010.01015.x.
Keoleian G.A., Volk T.A. (2005). Renewable energy from willow biomass crops: life cycle energy, environmental and economic performance. Critical Reviews in Plant Sciences 24(5–6): 385–406. https://doi.org/10.1080/07352680500316334.
Kim M.S., Richardson B.A., McDonald G.I., Klopfenstein N.B. (2011). Genetic diversity and structure of western white pine (Pinus monticola) in North America: a baseline study for conservation, restoration, and addressing impacts of climate change. Tree Genetics & Genomes 7(1): 11–21. https://doi.org/10.1007/s11295-010-0311-0.
Klang-Westin E., Perttu K. (2002). Effects of nutrient supply and soil cadmium concentration on cadmium removal by willow. Biomass Bioenergy 23(6): 415–426. https://doi.org/10.1016/S0961-9534(02)00068-5.
Kocik A., Truchan M., Rozen A. (2007). Application of willows (Salix viminalis) and earthworms (Eisenia fetida) in sewage sludge treatment. European Journal of Soil Biology 43(S1): S327–S331. https://doi.org/10.1016/j.ejsobi.2007.08.019.
Lamborot M., Eaton L. (1997). The Maipo River as a biogeographical barrier to Liolaemus monticola (Tropiduridae) in the mountain ranges of central Chile. Journal of Zoological Systematics and Evolutionary Research 35:105–111. https://doi.org/10.1111/j.1439-0469.1997.tb00411.x.
Lascoux M., Thorsén J., Gullberg U. (1996). Population structure of a riparian willow species, Salix viminalis L. Genetical Research 68(1): 45–54. https://doi.org/10.1017/S0016672300033875.
Lee K.M., Kim Y.Y., Hyun J.O. (2011). Genetic variation in populations of Populus davidiana Dode based on microsatellite marker analysis. Genes & Genomics 33(2): 163–171. https://doi.org/10.1007/s13258-010-0148-9.
Lian C., Nara K., Nakaya H., Zhou Z., Wu B., Miyashita N., Hogetsu T. (2001). Development of microsatellite markers in polyploid Salix reinii. Molecular Ecology Notes 1: 160–161. https://doi.org/10.1046/j.1471-8278.2001.00059.x.
Lin J., Gibbs J.P., Smart L.B. (2009). Population genetic structure of native versus naturalized sympatric shrub willows (Salix; Salicaceae). American Journal of Botany 96(4): 771–785. https://doi.org/10.3732/ajb.0800321.
Liu J., Zheng X., Potter D., Hu C., Teng Y. (2012). Genetic diversity and population structure of Pyrus calleryana (Rosaceae) in Zhejiang province, China. Biochemical Systematics and Ecology 45: 69–78. https://doi.org/10.1016/j.bse.2012.06.027.
Liu K., Muse S.V. (2005). PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21(9): 2128–2129. https://doi.org/10.1093/bioinformatics/bti282.
Mahdi J.G. (2010). Medicinal potential of willow: a chemical perspective of aspirin discovery. Journal of Saudi Chemical Society 14(3): 317–322. https://doi.org/10.1016/j.jscs.2010.04.010.
Mandel J.R., Dechaine J.M., Marek L.F., Burke J.M. (2011). Genetic diversity and population structure in cultivated sunflower and a comparison to its wild progenitor, Helianthus annuus L. Theoretical and Applied Genetics 123(5): 693–704. https://doi.org/10.1007/s00122-011-1619-3.
Manni F., Guerard E., Heyer E. (2004). Geographic patterns of (genetic, morphologic, linguistic) variation: how barriers can be detected by using Monmonier’s algorithm. Human Biology 76(2): 173–190. https://doi.org/10.1353/hub.2004.0034.
Mehes M., Nkongolo K.K., Michael P. (2009). Assessing genetic diversity and structure of fragmented populations of eastern white pine (Pinus strobus) and western white pine (P. monticola) for conservation management. Journal of Plant Ecology 2(3): 143–151. https://doi.org/10.1093/jpe/rtp016.
Millar C.I., Libby W.J. (1991). Strategies for conserving clinal, ecotypic, and disjunct population diversity in widespread species. In: Falk D.A., Holsinger K.E. (eds.). Genetics and conservation of rare plants. Oxford University Press, New York. p. 149–170.
Mleczek M., Rutkowski P., Rissmann I., Kaczmarek Z., Golinski P., Szentner K., Strazynska K., Stachowiak A. (2010). Biomass productivity and phytoremediation potential of Salix alba and Salix viminalis. Biomass Bioenergy 34(9): 1410–1418. https://doi.org/10.1016/j.biombioe.2010.04.012.
Newsholme C. (1992). Willows: the genus Salix, 2nd edn. Timber Press, Portland.
Peakall R., Smouse P.E. (2012). GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research – an update. Bioinformatics 28(19): 2537–2539. https://doi.org/10.1093/bioinformatics/bts460.
Perdereau A.C., Kelleher C.T., Douglas G.C., Hodkinson T.R. (2014). High levels of gene flow and genetic diversity in Irish populations of Salix caprea L. inferred from chloroplast and nuclear SSR markers. BMC Plant Biology 14(202). 12 p. https://doi.org/10.1186/s12870-014-0202-x.
Pritchard J.K., Stephens M., Donnelly P. (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Przyborowski J.A., Sulima P. (2010). The analysis of genetic diversity of Salix viminalis genotypes as a potential source of biomass by RAPD markers. Industrial Crops and Products 31(2): 395–400. https://doi.org/10.1016/j.indcrop.2009.12.009.
Rathmacher G., Niggemann M., Köhnen M., Ziegenhagen B., Bialozyt R. (2010). Short-distance gene flow in Populus nigra L. accounts for small-scale spatial genetic structures: implications for in situ conservation measures. Conservation Genetics 11(4): 1327–1338. https://doi.org/10.1007/s10592-009-9961-6.
Rönnberg-Wästljung A.C. (2001). Genetic structure of growth and phenological traits in Salix viminalis. Canadian Journal of Forest Research 31(2): 276–282. https://doi.org/10.1139/x00-175.
Shriver M., Jin L., Boerwinkle E., Deka R., Ferrell R.E., Chakraboryt R. (1995). A novel measure of genetic distance for highly polymorphic tandem repeat loci. Molecular Biology and Evolution 12(5): 914–920. https://doi.org/10.1093/oxfordjournals.molbev.a040268.
Smulders M.J.M., Cottrell J.E., Lefèvre F., van der Schoot J., Arens P., Vosman B., Tabbener F., Grassi T., Fossati S., Castiglione S., Krystufek V., Fluch S., Burg K., Vornam B., Pohl A., Gebhardt K., Alba N., Agúndez D., Maestro C., Notivol E., Volosyanchuk R., Pospíšková M., Bordács S., Bovenschen J., van Dam B.C., Koelewijn H.P., Halfmaerten D., Ivens B., van Slycken J., Vanden Broeck A., Storme V., Boerjan W. (2008). Structure of the genetic diversity in black poplar (Populus nigra L.) populations across European river systems: consequences for conservation and restoration. Forest Ecology and Management 255(5–6): 1388–1399. https://doi.org/10.1016/j.foreco.2007.10.063.
Sochor M., Vašut R.J., Bártová E., Majeský Ľ., Mráček J. (2013). Can gene flow among populations counteract the habitat loss of extremely fragile biotopes? An example from the population genetic structure in Salix daphnoides. Tree Genetics & Genomes 9(5): 1193–1205. https://doi.org/10.1007/s11295-013-0628-6.
Stamati K., Blackie S., Brown J.W.S., Russell J. (2003). A set of polymorphic SSR loci for subarctic willow (Salix lanata, S. lapponum and S. herbacea). Molecular Ecology Notes 3: 280–282. https://doi.org/10.1046/j.1471-8286.2003.00426.x.
Stamati K., Hollingsworth P.M., Russell J. (2007). Patterns of clonal diversity in three species of sub-arctic willow (Salix lanata, Salix lapponum and Salix herbacea). Plant Systematics and Evolution 269(1): 75–88. https://doi.org/10.1007/s00606-007-0578-2.
Talve T., McGlaughlin M.E., Helenurm K., Wallace L.E., Oja T. (2014). Population genetic diversity and species relationships in the genus Rhinanthus L. based on microsatellite markers. Plant Biology 16: 495–502. https://doi.org/10.1111/plb.12057.
Tamura K., Stecher G., Peterson D., Filipski A., Kumar S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Molecular Biology and Evolution 30(12): 2725–2729. https://doi.org/10.1093/molbev/mst197.
Teixeira H., Rodríguez-Echeverría S., Nabais C. (2014). Genetic diversity and differentiation of Juniperus thurifera in Spain and Morocco as determined by SSR. PloS One 9(2): e88996. https://doi.org/10.1371/journal.pone.0088996.
Trybush S.O., Jahodová Š., Čížková L., Karp A., Hanley S.J. (2012). High levels of genetic diversity in Salix viminalis of the Czech Republic as revealed by microsatellite markers. BioEnergy Research 5(4): 969–977. https://doi.org/10.1007/s12155-012-9212-4.
Wang L., Wang B.L., Wei Z.Z., Du Q.Z., Zhang D.Q., Li B.L. (2012). Development of 35 microsatellite markers from heat stress transcription factors in Populus simonii (Salicaceae). American Journal of Botany 0: e357–e361. https://doi.org/10.3732/ajb.1200056.
Wang P., Zhang M., Liu J., Xu L., Liu W. (2015). Genetic diversity and structure of Libanotis buchtormensis (Fisch.) DC. in disjunct populations along the bilateral sides of deserts in northwestern China. Plant Systematics and Evolution 301(9): 2219–2230. https://doi.org/10.1007/s00606-015-1225-y.
Watson C.C., Abt S.R., Derrick D. (1997). Willow posts bank stabilization. Journal of the American Water Resources Association 33: 293–300. https://doi.org/10.1111/j.1752-1688.1997.tb03510.x.
Wright S. (1978). Evolution and the genetics of populations: variability within and among natural populations. University of Chicago Press, Chicago.
Yeh F.C., Yang R.C., Boyle T.B.J., Ye Z.H., Mao J.X. (1997). POPGENE, the user-friendly shareware for population genetic analysis. Molecular biology and biotechnology centre, University of Alberta, Canada.
Yuan J., Cheng F.Y., Zhou S.L. (2012). Genetic structure of the tree peony (Paeonia rockii) and the Qinling Mountains as a geographic barrier driving the fragmentation of a large population. PLoS One 7(4): e34955. https://doi.org/10.1371/journal.pone.0034955.
Total of 62 references.
Send to email